Medicinski stručnjak članka

Nove publikacije
Usherov sindrom
Posljednji pregledao: 04.07.2025

Svi iLive sadržaji medicinski se pregledavaju ili provjeravaju kako bi se osigurala što je moguće točnija činjenica.
Imamo stroge smjernice za pronalaženje izvora i samo povezujemo s uglednim medijskim stranicama, akademskim istraživačkim institucijama i, kad god je to moguće, medicinski pregledanim studijama. Imajte na umu da su brojevi u zagradama ([1], [2], itd.) Poveznice koje se mogu kliknuti na ove studije.
Ako smatrate da je bilo koji od naših sadržaja netočan, zastario ili na neki drugi način upitan, odaberite ga i pritisnite Ctrl + Enter.
Usherov sindrom je nasljedna bolest koja se manifestira kao potpuna gluhoća od rođenja, kao i progresivna sljepoća s godinama. Gubitak vida povezan je s retinitis pigmentosa, procesom pigmentne degeneracije mrežnice. Mnogi ljudi s Usherovim sindromom također imaju teške probleme s ravnotežom.
Epidemiologija
Zahvaljujući istraživanju, bilo je moguće utvrditi da Usherov sindrom pogađa oko 8% pregledane gluhonijeme djece (testovi su provedeni u posebnim ustanovama za gluhonijeme osobe). Pigmentni retinitis uočen je kod 6-10% pacijenata koji pate od kongenitalne gluhoće, koja se pak uočava kod oko 30% osoba s pigmentnom bolešću mrežnice.
Vjeruje se da se ova bolest manifestira kod otprilike 3-10 ljudi od 100 tisuća diljem svijeta. Može se podjednako primijetiti i kod žena i kod muškaraca. Oko 5-6% svjetske populacije pati od ovog sindroma. Oko 10% svih slučajeva teške gluhoće u djetinjstvu javlja se zbog Usherovog sindroma I, kao i II tipa.
U Sjedinjenim Državama, tipovi 1 i 2 su najčešći tipovi. Zajedno čine otprilike 90 do 95 posto svih slučajeva Usherovog sindroma kod djece.
Uzroci Usherov sindrom
Usherov sindrom tipova I, II i III ima autosomno recesivni uzrok, dok se tip IV smatra poremećajem X-kromosoma. Uzroci sljepoće i gluhoće koji se javljaju kod ovog sindroma još nisu dovoljno proučeni. Pretpostavlja se da su osobe s ovom bolešću preosjetljive na komponente koje mogu oštetiti strukturu DNK. Osim toga, ova bolest može biti povezana s poremećajima imunološkog sustava, ali u ovom slučaju ne postoji točna slika tog procesa.
Godine 1989. kromosomske abnormalnosti prvi su put identificirane kod pacijenata s bolešću tipa II, što bi u budućnosti moglo dovesti do načina izolacije gena koji uzrokuju sindrom. Također bi moglo biti moguće identificirati te gene kod nositelja i razviti posebne prenatalne genetske testove.
 [ 8 ]
[ 8 ]
Faktori rizika
Sindrom se nasljeđuje kada su oba roditelja pogođena, tj. nasljeđuje se recesivnim tipom. Dijete također može naslijediti bolest ako su mu roditelji nositelji gena. Ako oba buduća roditelja imaju ovaj gen, tada je vjerojatnost da će imati dijete s ovim sindromom 1 prema 4. Osoba koja ima samo jedan gen za sindrom smatra se nositeljem, ali nema simptome poremećaja. Danas još nije moguće utvrditi ima li osoba gen za ovu bolest.
Ako se dijete rodi od roditelja, od kojih jedan nema takav gen, vjerojatnost da će naslijediti sindrom je vrlo niska, ali će definitivno biti nositelj.
Simptomi Usherov sindrom
Simptomi Usherovog sindroma uključuju gubitak sluha i abnormalno nakupljanje pigmentiranih stanica u očnim strukturama. Pacijent zatim razvija degeneraciju mrežnice, što uzrokuje pogoršanje vida i na kraju gubitak vida u najtežim slučajevima.
Senzorineuralni gubitak sluha može biti blag ili potpun i obično ne napreduje od rođenja. Međutim, bolest pigmenta mrežnice može se početi razvijati u djetinjstvu ili kasnije. Rezultati testova pokazali su da se centralna vidna oštrina može održati dugi niz godina, čak i kada se periferni vid pogorša (stanje koje se naziva "tunelski vid").
To su glavne manifestacije bolesti, koje ponekad mogu biti nadopunjene drugim poremećajima, poput psihoze i drugih mentalnih poremećaja, problema s unutarnjim uhom i/ili katarakte.
Obrasci
Tijekom istraživanja identificirane su 3 vrste ove bolesti, kao i 4. oblik koji je prilično rijedak.
Tip I bolesti karakterizira kongenitalna potpuna gluhoća, kao i poremećaj ravnoteže. Često takva djeca počinju hodati tek u dobi od 1,5 godina. Pogoršanje vida obično počinje u dobi od 10 godina, a konačni razvoj stanja noćne sljepoće počinje u dobi od 20 godina. Djeca s ovom vrstom bolesti mogu razviti progresivno pogoršanje perifernog vida.
Kod bolesti tipa II opaža se umjerena ili kongenitalna gluhoća. U tom slučaju, pogoršanje djelomične gluhoće često se više ne događa. Pigmentni retinitis počinje se razvijati oko kraja adolescencije ili nakon 20 godina. Razvoj noćne sljepoće obično počinje u dobi od 29-31 godine. Oštećenje vidne oštrine u slučaju patologije tipa II općenito napreduje nešto sporije nego kod tipa I.
Tip III bolesti karakterizira progresivni gubitak sluha, koji obično počinje tijekom puberteta, kao i postupni razvoj retinitis pigmentose u istom razdoblju (nešto kasnije od gubitka sluha), što može postati faktor u razvoju progresivne sljepoće.
Manifestacije patologije tipa IV uglavnom se javljaju kod muškaraca. U ovom slučaju se opažaju i progresivni poremećaji te gubitak sluha i vida. Ovaj oblik je vrlo rijedak i obično ima X-kromosomsku prirodu.
Dijagnostika Usherov sindrom
Dijagnoza Usherovog sindroma postavlja se na temelju pacijentove uočene kombinacije iznenadne gluhoće i progresivnog gubitka vida.
Testovi
Za otkrivanje mutacije može se naručiti poseban genetski test.
Pronađeno je jedanaest genetskih lokusa koji mogu uzrokovati razvoj Usherovog sindroma, a identificirano je devet gena koji su definitivno uzrok poremećaja:
- Tip 1: MY07A, USH1C, Cdh23, Pcdh15, SANS.
- Tip 2: ush2a, VLGR1, WHRN.
- Usherov sindrom tip 3: USH3A.
Znanstvenici NIDCD-a, zajedno s kolegama sa sveučilišta u New Yorku i Izraelu, identificirali su mutaciju nazvanu R245X u genu Pcdh15 koja je odgovorna za veliki postotak Usherovog sindroma tipa 1 u židovskoj populaciji.
Za informacije o laboratorijima koji provode klinička ispitivanja posjetite https://www.genetests.org i pretražite imenik laboratorija za "Usherov sindrom".
Za informacije o postojećim kliničkim ispitivanjima koja uključuju genetsko testiranje za Usherov sindrom posjetite https://www.clinicaltrials.gov i pretražite "Usherov sindrom" ili "genetsko testiranje Usherovog sindroma".
[ 25 ], [ 26 ], [ 27 ], [ 28 ], [ 29 ], [ 30 ]
Instrumentalna dijagnostika
Postoji nekoliko metoda instrumentalne dijagnostike:
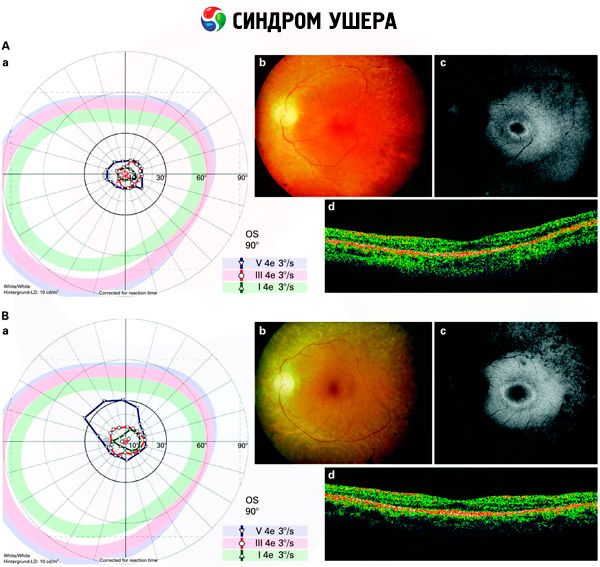
- Pregled fundusa kako bi se otkrila prisutnost pigmentnih mrlja na mrežnici, kao i sužavanje retinalnih žila;
- Elektroretinogram, koji omogućuje otkrivanje početnih degenerativnih odstupanja u mrežnici oka. Prikazuje izumiranje elektroradiografskih putova;
- Elektronistagmografija (ENG) mjeri nevoljne pokrete oka koji bi mogli ukazivati na prisutnost neravnoteže.
- Audiometrija, koja se koristi za utvrđivanje prisutnosti gluhoće i njezine težine.
Diferencijalna dijagnoza
Usherov sindrom treba razlikovati od nekih sličnih poremećaja.
Hallgrenov sindrom, koji karakterizira kongenitalni gubitak sluha i progresivni gubitak vida (razvijaju se i katarakta i nistagmus). Dodatni simptomi uključuju ataksiju, psihomotorne poremećaje, psihozu i mentalnu retardaciju.
Alstromov sindrom, nasljedna bolest u kojoj mrežnica degenerira, što rezultira gubitkom centralnog vida. Ovaj sindrom povezan je s pretilošću u djetinjstvu. Istovremeno, dijabetes melitus i gubitak sluha počinju se razvijati nakon 10 godina.
Rubeola kod trudnice u prvom tromjesečju može uzrokovati razne abnormalnosti u razvoju djeteta. Među posljedicama takve abnormalnosti su gubitak sluha, kao i (ili) problemi s vidom, a uz to i razni razvojni defekti.
Tko se može obratiti?
Liječenje Usherov sindrom
Trenutno ne postoji lijek za Usherov sindrom. Stoga se terapija u ovom slučaju uglavnom sastoji od usporavanja procesa gubitka vida, kao i kompenzacije gubitka sluha. Moguće metode liječenja uključuju:
- Uzimanje vitamina A (neki oftalmolozi vjeruju da visoke doze vitamina A palmitata mogu usporiti, ali ne i zaustaviti, napredovanje retinitis pigmentose);
- Ugradnja posebnih elektroničkih uređaja u pacijentove uši (slušni aparati, kohlearni implantati).
Oftalmolozi preporučuju da većina odraslih osoba s uobičajenim oblicima retinitis pigmentose dnevno uzima 15 000 IU (međunarodnih jedinica) vitamina A palmitata pod nadzorom. Budući da osobe s Usherovim sindromom tipa 1 nisu bile uključene u studiju, visoke doze vitamina A ne preporučuju se za ovu skupinu pacijenata. Osobe koje razmišljaju o uzimanju vitamina A trebale bi razgovarati o ovoj mogućnosti liječenja sa svojim liječnikom. Ostale preporuke za ovu mogućnost liječenja uključuju:
- Promijenite prehranu uključivanjem namirnica bogatih vitaminom A.
- Žene koje planiraju trudnoću trebaju prestati uzimati visoke doze vitamina A tri mjeseca prije planiranog začeća zbog povećanog rizika od urođenih mana.
- Žene koje su trudne trebaju prestati uzimati visoke doze vitamina A zbog povećanog rizika od urođenih mana.
Također je važno prilagoditi takvo dijete društvenom životu. To zahtijeva pomoć defektologa i psihologa. U slučaju da je pacijent počeo doživljavati progresivni gubitak vida, treba ga naučiti koristiti znakovni jezik.
Prognoza
Usherov sindrom ima nepovoljnu prognozu. Vidno polje i njegova oštrina počinju se pogoršavati u razdoblju od 20-30 godina kod većine pacijenata s ovom bolešću bilo koje vrste. U nekim slučajevima dolazi do potpunog bilateralnog gubitka vida. Gubitak sluha, koji je uvijek praćen nijemošću, vrlo se brzo razvija do potpunog bilateralnog gubitka sluha.