Medicinski stručnjak članka

Nove publikacije
Leahina subakutna nekrotizirajuća encefalomiopatija
Posljednji pregledao: 04.07.2025

Svi iLive sadržaji medicinski se pregledavaju ili provjeravaju kako bi se osigurala što je moguće točnija činjenica.
Imamo stroge smjernice za pronalaženje izvora i samo povezujemo s uglednim medijskim stranicama, akademskim istraživačkim institucijama i, kad god je to moguće, medicinski pregledanim studijama. Imajte na umu da su brojevi u zagradama ([1], [2], itd.) Poveznice koje se mogu kliknuti na ove studije.
Ako smatrate da je bilo koji od naših sadržaja netočan, zastario ili na neki drugi način upitan, odaberite ga i pritisnite Ctrl + Enter.
 [
[ Uzroci Leahovog sindroma
Bolest se temelji na nedostatku enzima koji osiguravaju proizvodnju energije uglavnom zbog poremećaja metabolizma piruvične kiseline i defekta u transportu elektrona u dišnom lancu. Razvija se nedostatak kompleksa piruvat dehidrogenaze (a-E1 podjedinica), piruvat karboksilaze, kompleksa 1 (NAD-koenzim Q-reduktaza) i kompleksa 4 (citokrom oksidaza) dišnog lanca.
Utvrđeno je da se defekti piruvat karboksilaze, kompleksa 1 (NAD-koenzim Q-reduktaza) i kompleksa 4 (citokrom oksidaza) dišnog lanca nasljeđuju autosomno recesivnim putem, defekti kompleksa piruvat dehidrogenaze (a-E1 podjedinica) nasljeđuju se X-vezano recesivnim putem. U slučaju točkastih mutacija mtDNA, koje utječu na 6. podjedinicu ATPaze, tipično je mitohondrijsko nasljeđivanje. Najčešće se javlja miscens mutacija, povezana sa zamjenom timina gvaninom ili citozinom na poziciji 8993 mtDNA. Rjeđa je mutacija na poziciji 9176 mtDNA. Zbog činjenice da je mutacija T8993G glavni defekt u NARP sindromu, opisane su obitelji s ove dvije bolesti. Kod djece je opisana i mutacija u mtDNA na poziciji 8344, koja se javlja kod MERRF sindroma.
Pretpostavlja se da se u slučaju nakupljanja mutirane mtDNA u većini mitohondrija razvija teški tijek Leighovog sindroma. U mitohondrijskom nastanku ovog stanja, mutirana mtDNA nalazi se u 90% svih mitohondrija. Patogeneza je povezana s kršenjem stvaranja energije u stanicama i razvojem laktične acidoze.
Simptomi Leahovog sindroma
Prvi znakovi bolesti pojavljuju se u ranoj dobi (1-3 godine). Međutim, poznati su slučajevi manifestacije bolesti u dobi od 2 tjedna i 6-7 godina. U početku se razvijaju nespecifični poremećaji: usporeni psihomotorni razvoj, smanjen apetit, epizode povraćanja, deficit tjelesne težine. Nakon toga se pojačavaju neurološki simptomi: mišićna hipotonija ili distonija s prijelazom u hipertoniju, napadi mioklonusa ili toničko-kloničkih napadaja, tremor ekstremiteta, koreoatetoza, poremećaj koordinacije, smanjeni tetivni refleksi, letargija, pospanost. Cerebralna neurodegeneracija je progresivna. Pojačavaju se simptomi piramidalne i ekstrapiramidalne insuficijencije, poremećen je akt gutanja. Često se opažaju promjene u organu vida poput ptoze, oftalmoplegije, atrofije vidnih živaca, rjeđe pigmentne degeneracije mrežnice. Ponekad se razvija hipertrofična kardiomiopatija, pojavljuju se epizode tahipneje.
Rijetko se bolest odvija kao akutna encefalopatija. Tipičniji je kronični ili subakutni tijek, koji dovodi do smrtnog ishoda nekoliko godina nakon početka bolesti. Kod brzog tijeka (nekoliko tjedana) smrt nastupa kao posljedica paralize respiratornog centra.
Dijagnostika Leahovog sindroma
Biokemijski test krvi otkriva laktacidozu zbog nakupljanja mliječne i piruvične kiseline u krvi i cerebrospinalnoj tekućini, kao i povećanje sadržaja alanina u krvi. Razina ketonskih tijela također može biti povišena. U urinu se otkriva povećano izlučivanje organskih kiselina: mliječne, fumarne itd. Razina karnitina u krvi i tkivima često se smanjuje.
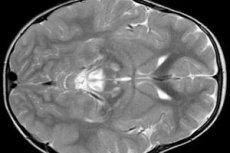
EEG rezultati otkrivaju fokalne znakove epileptičke aktivnosti. MRI podaci otkrivaju povećanje moždanih ventrikula, bilateralno oštećenje mozga, kalcifikaciju bazalnih ganglija (nucleus caudatus, putamen, substantia nigra, globus pallidus). Može se otkriti i atrofija moždanih hemisfera i moždane tvari.
Morfološki pregled otkriva grube promjene u moždanoj tvari: simetrična žarišta nekroze, demijelinizacije i spužvaste degeneracije mozga, uglavnom srednjih dijelova, ponsa, bazalnih ganglija, talamusa i vidnog živca. Histološka slika uključuje cističnu degeneraciju moždanog tkiva, astrocitnu gliozu, neuronsku smrt i povećanje broja mitohondrija u stanicama. U skeletnim mišićima dolazi do nakupljanja lipidnih inkluzija, smanjenja histokemijske reakcije na komplekse 1 i 4 respiratornog lanca, subsarkolemalnog nakupljanja mitohondrija, abnormalnih mitohondrija s dezorganizacijom krista. Fenomen RRF-a često se ne otkriva.
Kako ispitati?
Koji su testovi potrebni?
Использованная литература