Medicinski stručnjak članka

Nove publikacije
Alkaptonurija je kongenitalna abnormalnost enzima
Posljednji pregledao: 04.07.2025

Svi iLive sadržaji medicinski se pregledavaju ili provjeravaju kako bi se osigurala što je moguće točnija činjenica.
Imamo stroge smjernice za pronalaženje izvora i samo povezujemo s uglednim medijskim stranicama, akademskim istraživačkim institucijama i, kad god je to moguće, medicinski pregledanim studijama. Imajte na umu da su brojevi u zagradama ([1], [2], itd.) Poveznice koje se mogu kliknuti na ove studije.
Ako smatrate da je bilo koji od naših sadržaja netočan, zastario ili na neki drugi način upitan, odaberite ga i pritisnite Ctrl + Enter.
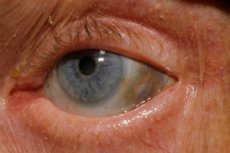
Jedan od vrlo rijetkih metaboličkih poremećaja, alkaptonurija, odnosi se na kongenitalne anomalije u metabolizmu aminokiseline tirozina.
Ovaj sindrom se može nazvati i nedostatkom homogentisat oksidaze, homogentisinurijom, nasljednom ohronozom ili bolešću crnog urina.[ 1 ]
Epidemiologija
Prema statistikama, nema više od devet slučajeva alkaptonurije na milijun ljudi. A u većini europskih zemalja postoji jedan slučaj na 100-250 tisuća živorođene djece.
Među europskim zemljama, iznimka je Slovačka (posebno relativno mala sjeverozapadna regija), gdje je prevalencija alkaptonurije jedan slučaj na 19 000 novorođenčadi. To je najvjerojatnije zbog činjenice da je među slovačkim romskim obiteljima koje tamo žive razina inbreedinga (brakova između rođaka) najviša u Europi: 10-14%. [ 2 ]
Uzroci alkaptonurija
Točni uzroci alkaptonurije, kao kongenitalnog poremećaja katabolizma (metaboličke razgradnje) aromatske (homocikličke) α-aminokiseline tirozina, utvrđeni su: ova vrsta metaboličkog poremećaja posljedica je homozigotnih ili složenih heterozigotnih mutacija jednog od tisuća gena na kromosomu 3, točnije, gena HGD na lokusu 3q21-q23 na dugom kraku kromosoma. Ovaj gen kodira nukleotidne sekvence jetrenog enzima homogentisat-1,2-dioksigenaze [ 3 ] (također nazvane homogentizinska kiselinska oksidaza ili homogentisat oksidaza) - metaloproteina koji sadrži željezo, potrebnog za jednu od faza razgradnje tirozina u tijelu. [ 4 ], [ 5 ]
Dakle, alkaptonurija je defekt enzima homogentisat-1,2-dioksigenaze, ili preciznije, rezultat njegovog genetski određenog nedostatka ili potpune odsutnosti. [ 6 ]
Budući da je alkaptonurija kongenitalni nedostatak enzima, nasljeđuje se autosomno recesivno, odnosno da bi se alkaptonurija pojavila kod djece, oba roditelja moraju imati modificirani gen za enzim, budući da svaki od njih djetetu prenosi samo jednu kopiju gena od dvije dostupne.
Prema najnovijim podacima, postoji više od dvjesto varijanti modifikacije HGD gena, a najčešće se opažaju missense mutacije, translokacija i splicing.
Faktori rizika
Jedini faktor rizika za razvoj ove kongenitalne enzimopatije je njezina prisutnost u obiteljskoj anamnezi i nasljeđivanje dvije modificirane kopije gena HGD, ako roditelji ne pokazuju alkaptonuriju (rizik prijenosa anomalije je 25%) ili jedan od roditelja ima ovaj poremećaj. [ 7 ]
Patogeneza
Tirozin igra ključnu ulogu u sintezi proteina, proizvodnji kromoproteina - pigmenta kože melanina, kao i hormona štitnjače i neurotransmitera kateholamina.
Mehanizam regulacije količine tirozina u stanicama je vrlo složen, a tijelo normalizira njegov višak sadržaja razgradnjom. Proces katabolizma tirozina, kao i svih aromatskih aminokiselina, je višestupanjski i odvija se u nekoliko faza. Svaka faza metaboličke razgradnje tirozina odvija se uz sudjelovanje specifičnog enzima i stvaranje međuprodukta.
Dakle, prvo se aminokiselina razgrađuje na para-hidroksifenilpiruvat, koji se pretvara u alkapton – 2,5-dihidroksifeniloctenu ili homogentizinsku kiselinu. Zatim bi se alkapton trebao transformirati u maleoctenu kiselinu, ali to se ne događa. [ 8 ]
A patogeneza alkaptonurije sastoji se od prestanka biokemijskih reakcija katabolizma tirozina u fazi stvaranja homogentizinske kiseline: jednostavno nema enzima potrebnog za njezinu razgradnju - homogentizat oksidaze.
Tijelo ne koristi homogentizinsku kiselinu i može se akumulirati izlučivanjem putem bubrega. Osim toga, oksidira se u benzokinoacetat (benzokinonooctena kiselina) koji, vezanjem na molekule tkiva i tjelesnih tekućina, tvori biopolimerne spojeve obojene poput melanina.
Nakupljanje ovih međuprodukata u tkivu dovodi do poremećaja kolagene strukture hrskavičnog tkiva, što smanjuje njegovu elastičnost – s pojavom mnogih kliničkih znakova alkaptonurije i razvojem komplikacija.
Simptomi alkaptonurija
Alkaptonurija kod novorođenčadi i dojenčadi karakterizirana je potamnjenjem urina. Kada je izložen zraku, urin na pelenama, pelenama i donjem rublju postaje tamnosmeđ; to je zbog nakupljanja i oslobađanja homogentizinske kiseline, koja se oksidira u benzokinoacetat. [ 9 ]
U nedostatku drugih simptoma, alkaptonurija kod male djece često se ne prepoznaje na vrijeme, budući da urin može potamniti nakon nekoliko sati mokrenja. Prema nekim podacima, samo petina djece mlađe od 12 mjeseci koja su rođena s nedostatkom ovog enzima identificira se u kliničkim uvjetima. Stoga je vrlo važno da roditelji obrate pozornost na brigu o svojoj dojenčadi.
Osim toga, rani znakovi uključuju pigmentaciju (plavičasto-siva boja) bjeloočnice i hrskavice ušiju i nosa, što se često naziva ohronoza.[ 10 ]
Vremenom se pojavljuju i drugi simptomi:
- jaka pigmentacija kože na jagodicama, pazusima i genitalijama;
- mrljanje odjeće pri kontaktu sa znojnim dijelovima tijela;
- napadi opće slabosti;
- promuklim glasom.
Treba imati na umu da su alkaptonurija i ohronoza, kao što je gore navedeno, sinonimi za isti poremećaj katabolizma tirozina.
Bolest javorovog sirupa urina i alkaptonurija. Kongenitalna bolest javorovog sirupa urina ili leucinoza također je metabolički poremećaj, ima isti obrazac nasljeđivanja, pa čak se i mutacije javljaju na istom kromosomu, ali utječu na gen koji kodira enzimski kompleks razgranatog lanca α-ketokiseline dehidrogenaze. Zbog toga tijelo ne može razgraditi određene komponente proteina, posebno aminokiseline leucin, izoleucin i valin. Kod ove bolesti, urin (i ušni vosak) imaju sladak miris; osim toga, klinička slika ove vrste organske acidemije uključuje hipopigmentaciju, fluktuacije krvnog tlaka, napadaje, povraćanje i proljev, pad razine glukoze u krvi, ketoacidozu, halucinacije itd. Stopa smrtnosti kod djece je prilično visoka; kod odraslih, bez liječenja, može doći do kome i smrti zbog cerebralnog edema.
Albinizam i alkaptonuriju „ujedinuje“ samo tirozin. Albinizam, uključujući okulokutani, uzrokovan je genetskim mutacijama koje utječu na proizvodnju pigmenta melanina. Kongenitalne promjene uočene su u genu TYR na kromosomu 11 (11q14.3), koji kodira tirozinazu, melanosomski enzim koji sadrži bakar, neophodan za stvaranje pigmenta kože na temelju produkata metabolizma tirozina. Ova bolest je mnogo češća od alkaptonurije.
Komplikacije i posljedice
Uzrokovana djelovanjem međuproduktnih metabolita tirozina - homogentizinske i benzokinonoacetatne kiseline - posljedice i komplikacije alkaptonurije javljaju se zbog taloženja reaktivnih pigmentiranih polimera, uništavanja kolagenih fibrila i pogoršanja stanja hrskavice (uz smanjenje njihove otpornosti na mehanički stres).
Tijekom godina, u odrasloj dobi, razvijaju se degenerativni artritis i osteoartritis velikih zglobova (kuka, sakroilijakalnog i koljena); intervertebralni prostori se sužavaju (osobito u lumbalnoj i torakalnoj kralježnici) - s kalcifikacijom i stvaranjem osteofita; gustoća tkiva subhondralnih koštanih ploča se smanjuje, a temeljne kosti mogu proći patološko preoblikovanje s nastankom izraslina i deformacija. [ 11 ]
Može se uočiti oštećenje srčanih zalistaka (aortnog i mitralnog) i koronarnih arterija – sa znakovima koronarne bolesti srca, kao i stvaranje kamenaca u bubrezima i prostati – zbog iste kalcifikacije. [ 12 ], [ 13 ]
Dijagnostika alkaptonurija
Tipično, dijagnoza kongenitalnih metaboličkih poremećaja temelji se na proučavanju bioloških tekućina u tijelu.
Na temelju kojih testova i reakcija se može dijagnosticirati alkaptonurija? Potrebni su testovi urina za otkrivanje homogentizinske kiseline i određivanje njezine razine (normalna – 20-30 mg dnevno, povišena – 3-8 g). Uzorak urina pregledava se plinskom kromatografijom ili masenom spektrometrijom, primjenom tekućinske kromatografije; moguć je probirni test na prisutnost željezovog klorida u urinu. [ 14 ]
Postoji i metoda za brzu dijagnostiku – određivanje alkaptona u osušenim mrljama urina na papiru (intenzitetom boje).
Prilikom razjašnjavanja dijagnoze, instrumentalna dijagnostika (radiografija) uključuje identificiranje radioloških znakova osteoartritisa i drugih patologija zglobova kod pacijenata.
Dijagnoza se potvrđuje molekularno-genetskim metodama dijagnosticiranja nasljednih bolesti, kao što su genetsko testiranje i sekvenciranje DNK. [ 15 ]
Diferencijalna dijagnoza
Diferencijalna dijagnoza uključuje hemokromatozu i akutno zatajenje jetre novorođenčeta, melaninuriju, akutnu intermitentnu porfiriju, hemofagocitnu limfohistiocitozu, primarnu mitohondrijalnu patologiju, reumatoidni artritis, ankilozantni spondilitis.
Tko se može obratiti?
Liječenje alkaptonurija
Glavni tretman za alkaptonuriju je oralna primjena velikih doza (najmanje 1000 mg dnevno) askorbinske kiseline. Kod djece to povećava izlučivanje homogentizinske kiseline u urinu, a kod odraslih smanjuje sadržaj u urinu njezinog derivata, benzokinonooctene kiseline, i usporava njezino vezanje za strukture vezivnog tkiva zglobova i kolagena. [ 16 ]
Zapadnoeuropske klinike testiraju lijek Nitizinon (Orfalin), lijek iz skupine metabolita koji inhibira drugu fazu katabolizma tirozina: transformaciju para-hidroksifenilpiruvata u homogentizinsku kiselinu. Međutim, primjena ovog farmakološkog sredstva dovodi do nakupljanja tirozina i može uzrokovati teške nuspojave, uključujući zamućenje rožnice i fotofobiju, krvarenje iz nosa i želuca, zatajenje jetre, promjene u krvi itd. Ipak, u Sjedinjenim Državama Nitizinon je odobren od strane FDA za liječenje tirozinemije tipa I. [17 ], [ 18 ]
Stoga se za probleme sa zglobovima uzrokovane alkaptonurijom provodi fizioterapija – terapija vježbanjem za povećanje mišićne snage i poboljšanje pokretljivosti zglobova, balneoterapija i peloidna terapija za ograničavanje boli.
Iako se tirozin ne unosi samo hranom, već se i proizvodi u tijelu, pacijentima s alkaptonurijom preporučuje se dijeta s niskim udjelom proteina i ograničavanje konzumacije hrane bogate tirozinom, prvenstveno govedine i svinjetine, mliječnih proizvoda (posebno sireva), mahunarki, orašastih plodova i sjemenki.
Prevencija
Sprječavanje genskih mutacija je nemoguće, ali kako bi se spriječilo rađanje djece s visokim rizikom od kongenitalnih poremećaja, postoji medicinsko-genetsko savjetovanje, koje je potrebno prije planirane trudnoće za one parove čija obiteljska anamneza uključuje nasljedne bolesti. [ 19 ]
Prognoza
Smrtni ishodi alkaptonurije su vrlo rijetki, a smrt može biti uzrokovana ozbiljnim komplikacijama koje uključuju srce i bubrege. Dakle, ukupni životni vijek ljudi s alkaptonurijom je dobar.
Ali kvaliteta života je smanjena zbog intenzivne boli u zglobovima ili kralježnici sa značajnim ograničenjem pokretljivosti, često progresivne.