
Vrsta 3 mukopolisaharidoze
Posljednji pregledao: 23.04.2024

Svi iLive sadržaji medicinski se pregledavaju ili provjeravaju kako bi se osigurala što je moguće točnija činjenica.
Imamo stroge smjernice za pronalaženje izvora i samo povezujemo s uglednim medijskim stranicama, akademskim istraživačkim institucijama i, kad god je to moguće, medicinski pregledanim studijama. Imajte na umu da su brojevi u zagradama ([1], [2], itd.) Poveznice koje se mogu kliknuti na ove studije.
Ako smatrate da je bilo koji od naših sadržaja netočan, zastario ili na neki drugi način upitan, odaberite ga i pritisnite Ctrl + Enter.
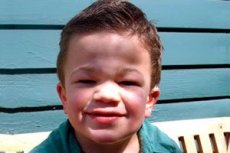
Mucopolysaccharidosis tipa III (sinonimi: Sanfilippo sindrom, zatajenje lizosomskog aN-acetylglucosaminidase - mucopolysaccharidosis Sh A, acetil-CoA i-glukosaminid-N-acetiltransferaze - mucopolysaccharidosis III B, N-acetilglukozamin-6-sulfataza - mucopolysaccharidosis III C, sulfamidazy - mucopolysaccharidose III D).
 [
[Patogeneza
Od bolesti uzrokuju mutacije četiri različita gena: lizosomalnu AN-acetylglucosaminidase (mucopolysaccharidosis III A), acetil-CoA i-glukosaminid-N-acetiltransferaze (mucopolysaccharidosis III B), N-acetilglukozamin lizosomskog-6-sulfataze (III mucopolysaccharidosis C) sulfamidazy (mucopolysaccharidosis III D). Svi enzimi su uključeni u metabolizam heparan sulfata.
Heparan-N-sulfataza gen-SGSH-nalazi se na dugoj ruci kromosoma 17-17q25.3. 75,3% trenutno poznatih mutacija u SGSH genu su točke mutacija. Česte mutacije tipične za europske populacije - R74C (56% u Poljskoj i 21% u Njemačkoj) i R245H (56% u Nizozemskoj).
Frekvencija mutacije R74C je 47,5%, mutacija R245H je 7,5%. Dvije druge mutacije, delll35G i N389S, zajedno čine 21,7% mutiranih alela.
Gen n-acetilglukozaminidaza, NAGLU, nalazi se na dugoj ruci kromosoma 17-17q21. 69% mutacija pronađenih u NAGLU genu je manijak i glupost mutacija, 26,3% su male delecije i umetanja. Gen, acetil-CoA-cc-glukozaminid-N-acetiltransferaza, HGSNAT, nalazi se na kratkom kraku kromosoma 8-8p11.1. Genet je karakteriziran samo u 2006 i do danas je pronašao samo nekoliko mutacija.
Genet N-acetil-glukozamin-6-sulfataza - GNS - nalazi se na dugoj ruci kromosoma 12 - 12ql4. U svijetu je zabilježeno 12 bolesnika s mukopolisaharidozom IIID. Postoje 4 mutacije u GNS genu.
Kada su svi podtipa III mucopolysaccharidosis povrede javlja razgradnju heparan sulfata uključena je u strukturi stanične membrane, uključujući membrane neurona koji se povezuje s ozbiljnim neurodegenerativnih procesa koji su uzrokovani kortikalnom atrofijom. Kronična dijareja pripisuje uključen u patološke procesu autonomnog nervnog sistema zajedno s disfunkcijom intestinalne mukoze. Senzorineuralna gubitak sluha je vjerojatno zbog tri razloga: česte upale srednjeg uha, uho kosti deformitetima i abnormalnosti unutarnjeg uha. Krutost zglobova rezultat je deformacije metafizika, zadebljanje zglobne kapsule je sekundarno od taloženja glikozaminoglikana i fibroze u njemu. Intrasyndromic razlike u težini bolesti su jedino posljedica preostale funkcionalne aktivnosti mutantnog enzima: što je veći, lakše napreduje bolest.
Simptomi vrsta 3 mukopolisaharidoze
Klinički polimorfizam u sindromu Sanfilippa je manje izražen nego kod drugih vrsta mukopolisaharidoze. Karakteriziran sporom progresijom bolesti, teškim neurološkim poremećajima s teškim simptomima iz unutarnjih organa i drugih sustava.
Prvi simptomi bolesti obično se pojavljuju u dobi od 2-6 godina kod djece s prethodnim normalnim razvojem. Manifestni simptomi uključuju regresiju razvoja psihomotora i govora, psihijatrijskih poremećaja u obliku razvoja sindroma hiperaktivnosti, autističnog ili agresivnog ponašanja, poremećaja spavanja; djeca postaju neoprezna i nepažljiva.
Drugi uobičajeni simptomi su hirsutizam, tvrda kosa, blaga hepatosplenomegalija, deformacija valgija ekstremiteta, kratki vrat. Formiranje grubih crta lica prema vrsti gargoilizma i skeletnih deformacija višestruko dysostosis slabo izražena na Mucopolysaccharidosis III u usporedbi s drugim vrstama, naznačen mucopolysaccharidosis Hurler fenotipa. Rast, u pravilu, odgovara dobi, a zglobna krutost rijetko uzrokuje kršenje njihovih funkcija. Većina bolesnika često razvija osteoporozu i osteomalazu. Sekundarni skeletni poremećaji visoki su rizik patoloških prijeloma. Najčešći psionurološki poremećaji najčešće se promatraju 6-10 godina života, dovode do teških socijalnih nepodudarnosti. Progresivni neurosensorni gubitak sluha je svojstveno svim bolesnicima s teškim i umjerenim oblicima bolesti. Konvulzije se opažaju u gotovo svim bolesnicima dok bolest napreduje.
Tijek bolesti napreduje brzo, većina pacijenata ne preživi do dobi od 20 godina. Vjeruje se da je mukopolisaharidoza IIIA najčešći i najteži tip ovog sindroma.
Dijagnostika vrsta 3 mukopolisaharidoze
Dijagnoza mukopolisaharidoze III potvrđena je određivanjem razine izlučivanja glukozamicina iz mokraćnog mjehura i mjerenjem enzimske aktivnosti. U slucaju mukopolisaharidoze III, ukupni izlučivanje glikozaminoglikana u mokraći se povećava i opaža se hipereekskricija heparan sulfata. Aktivnost lizosomskog enzima koji odgovara određenom podtipu mukopolisaharidoze III mjeri se u leukocitima ili kulturi kožnih fibroblasta pomoću umjetnog fluorogenskog supstrata.
Dijagnostika moguće su mjerenjem enzimske aktivnosti u korionskog villus biopsiju na 9-11 tjedna trudnoće i / ili utvrđivanje spektra glikozaminoglikana u amnionske tekućine na 20-22 tjedna trudnoće. Za obitelji s poznatim genotipom, moguće je provesti DNA dijagnostiku u ranoj fazi trudnoće.
Koji su testovi potrebni?
Diferencijalna dijagnoza
Diferencijalna dijagnoza se provodi u Mucopolysaccharidosis skupine, a s druge poremećaje lizosoma skladištenja: Mucolipidosis, galaktosialidozom, sialidosis, mannozidozom, fucosidosis, GM1-gangliosidosis.
Tko se može obratiti?
Liječenje vrsta 3 mukopolisaharidoze
Do danas nisu razvijene učinkovite terapije za mucopolysaccharidosis III. Označena je simptomatska terapija.
Использованная литература