Medicinski stručnjak članka

Alkaptonurija - kongenitalna enzimska patologija
Posljednji pregledao: 23.04.2024

Svi iLive sadržaji medicinski se pregledavaju ili provjeravaju kako bi se osigurala što je moguće točnija činjenica.
Imamo stroge smjernice za pronalaženje izvora i samo povezujemo s uglednim medijskim stranicama, akademskim istraživačkim institucijama i, kad god je to moguće, medicinski pregledanim studijama. Imajte na umu da su brojevi u zagradama ([1], [2], itd.) Poveznice koje se mogu kliknuti na ove studije.
Ako smatrate da je bilo koji od naših sadržaja netočan, zastario ili na neki drugi način upitan, odaberite ga i pritisnite Ctrl + Enter.
Jedan od vrlo rijetkih metaboličkih poremećaja - alkaptonurija - odnosi se na kongenitalne abnormalnosti u metabolizmu aminokiseline tirozin.
Također, ovaj sindrom se može nazvati nedostatkom homogentisat oksidaze, homogentisinurijom, nasljednom ohronozom ili bolešću crnog urina. [1]
Epidemiologija
Prema statistikama, ne postoji više od devet slučajeva alkaptonurije na 1 milijun stanovništva. A u većini europskih zemalja - jedan slučaj na 100-250 tisuća živorođene djece.
Među europskim zemljama iznimka je Slovačka (osobito relativno mala sjeverozapadna regija), gdje je prevalencija alkaptonurije jedan slučaj na 19 tisuća novorođenčadi. Po svoj prilici, to je posljedica činjenice da je među obiteljima slovačkih Roma koji tamo žive, razina inbreedinga (brakovi između rođaka) najviša u Europi: 10-14%. [2]
Uzroci alkaptonurija
Utvrđeni su točni uzroci alkaptonurije, kao urođenog poremećaja katabolizma (metaboličkog razgradnje) aromatske (homociklične) α-aminokiseline tirozina: ova vrsta metaboličkog poremećaja posljedica je homozigotnih ili složenih heterozigotnih mutacija jedne od tisuća gena na kromosomu 3, točnije gen HqG21 u lokusu 3 -q23 na dugom kraku kromosoma. Ovaj gen kodira nukleotidne sekvence enzima jetre homogentisat-1,2-dioksigenaze [3](koja se također naziva i oksidaza homogentizinske kiseline ili homogentisat oksidaza), metaloproteina koji sadrži željezo, koji je potreban za jednu od faza razgradnje tirozina u tijelu. [4], [5]
Dakle, alkaptonurija je nedostatak enzima homogentisat-1,2-dioksigenaze, točnije posljedica njezina genetski uvjetovanog nedostatka ili potpunog odsustva. [6]
Kao urođena fermentopatija, alkaptonurija se nasljeđuje kao autosomno recesivno svojstvo, odnosno da bi se alkaptonurija javila kod djece, oba roditelja moraju imati modificirani gen za ovaj enzim, jer svaki od njih prenosi djetetu samo jednu kopiju gen iz dva dostupna.
Prema posljednjim podacima, postoji više od dvjesto varijanti modifikacije gena HGD, a najčešće se promatraju missense mutacije, translokacija i spajanje.
Faktori rizika
Jedini čimbenik rizika za razvoj ove kongenitalne fermentopatije je njezina obiteljska anamneza i nasljeđivanje dvije modificirane kopije gena HGD, ako roditelji ne pokazuju alkaptonuriju (rizik prijenosa anomalije je 25%), ili jednu od roditelji imaju ovaj poremećaj. [7]
Patogeneza
Najvažniju ulogu tirozin ima u sintezi proteina, proizvodnji kromoproteina - pigmenta kože melanina, kao i hormona štitnjače i neurotransmitera katekolamina.
Mehanizam za regulaciju količine tirozina u stanicama vrlo je složen, a tijelo cijepanjem normalizira svoj višak sadržaja. Proces katabolizma tirozina, kao i svih aromatskih aminokiselina, je višestepeni i odvija se u nekoliko faza. Štoviše, svaka faza metaboličkog razgradnje tirozina odvija se uz sudjelovanje određenog enzima i stvaranje međuproizvoda.
Dakle, prvo se aminokiselina cijepa na para-hidroksifenilpiruvat, koji se pretvara u alkapon-2,5-dihidroksifenilactenu ili homogentisatnu kiselinu. Nadalje, alkapon bi trebao biti pretvoren u maleoctenu kiselinu, ali to se ne događa. [8]
Patogeneza alkaptonurije sastoji se u zaustavljanju biokemijskih reakcija katabolizma tirozina u fazi stvaranja homogentizinske kiseline: jednostavno ne postoji potreban enzim za njezinu razgradnju - homogentisat oksidaza.
Homogentizinska kiselina se ne koristi u tijelu i može se akumulirati izlučivanjem kroz bubrege. Osim toga, oksidira se u benzokinoacetat (benzokinoctene kiseline), koji vezanjem s molekulama tkiva i tjelesnih tekućina tvori biopolimerne spojeve obojene poput melanina.
Nakupljanje ovih međuproizvoda u tkivu dovodi do poremećaja strukture kolagena u hrskavičnom tkivu, zbog čega se smanjuje njegova elastičnost - s pojavom mnogih kliničkih znakova alkaptonurije i razvojem komplikacija.
Simptomi alkaptonurija
Alkaptonurija u novorođenčadi i dojenčadi očituje se potamnjenjem urina. U dodiru sa zrakom, boja urina na pelenama, pelenama i donjem rublju postaje tamnosmeđa; to je posljedica nakupljanja i oslobađanja homogentisatne kiseline koja se oksidira u benzokinoacetat. [9]
U nedostatku drugih simptoma, alkaptonurija u djece u ranoj dobi često se ne prepoznaje pravodobno, jer nakon mokrenja urin može potamniti nakon nekoliko sati. Prema nekim izvješćima, u kliničkim uvjetima otkrivena je samo jedna petina djece mlađe od 12 mjeseci koja su rođena s ovom fermentopatijom. Stoga je vrlo važno da roditelji obrate pozornost na brigu o bebi.
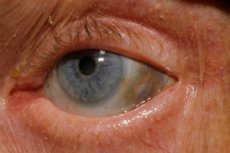
Osim toga, rani znakovi uključuju pigmentaciju (plavkasto sivu) očne bjeloočnice i hrskavice ušnih školjki i nosa, često nazivanu ohronozom. [10]
S vremenom se pojavljuju i drugi simptomi:
- jaka pigmentacija kože na jagodicama, pazuhu i genitalijama;
- bojanje odjeće u dodiru s znojnim dijelovima tijela;
- napadi opće slabosti;
- promukli glas.
Treba imati na umu da alkaptonuria i ochronosis , kao što je gore navedeno, su imena sinonima za istu povredu tirozin katabolizma.
Bolest javorovog sirupa i alkaptonurija. Urođena bolest javorovog sirupa ili leucinoza također se odnosi na metaboličke poremećaje, ima istu vrstu nasljeđivanja, pa se čak i mutacije javljaju na istom kromosomu, ali se odnose na gen koji kodira enzimski kompleks razgranate α-keto kiseline dehidrogenaze. Zbog toga tijelo ne može razgraditi određene proteinske komponente, osobito aminokiseline leucin, izoleucin i valin. U tom stanju urin (i ušni vosak) imaju slatkast miris; osim toga, u kliničkoj slici ove vrste organske acidemije opaža se hipopigmentacija, fluktuacije krvnog tlaka, konvulzije, povraćanje i proljev, pad razine glukoze u krvi, ketoacidoza, halucinacije itd. Stopa smrtnosti u djece prilično je velika visoka; u odraslih osoba bez liječenja može doći do kome i smrti zbog cerebralnog edema.
Albinizam i alkaptonurija "ujedinjuju" samo tirozin. Albinizam , uključujući okulokutani albinizam, uzrokovan je genetskim mutacijama koje utječu na proizvodnju pigmenta melanina. Urođene promjene zabilježene su u TYR genu na kromosomu 11 (11q14.3), koji kodira tirozinazu, enzim koji sadrži bakar u melanosomima koji je neophodan za stvaranje pigmenta kože temeljenog na produktima metabolizma tirozina. Ova je bolest mnogo češća od alkaptonurije.
Komplikacije i posljedice
Posljedice i komplikacije alkaptonurije uzrokovane djelovanjem posrednih metabolita tirozina - homogentizinske i benzokinoctene kiseline - pojavljuju se zbog taloženja reaktivnih pigmentiranih polimera, uništavanja kolagenskih vlakana i pogoršanja stanja hrskavice (sa smanjenjem njihove rezistencije) na mehaničko naprezanje).
Godinama kasnije, u odrasloj dobi, razvijaju se degenerativni artritis i osteoartritis velikih zglobova (kuka, sakroilijaka i koljena); sužavanje intervertebralnih prostora (osobito lumbalne i torakalne kralježnice) - s kalcifikacijom i stvaranjem osteofita; gustoća tkiva subhondralnih koštanih ploča smanjuje se, a kosti ispod njih mogu proći patološko remodeliranje s stvaranjem izraslina i deformacija. [11]
Može doći do oštećenja srčanih zalistaka (aortnih i mitralnih) i koronarnih arterija, sa znakovima koronarne bolesti srca, kao i stvaranja kamena u bubrezima i prostati, zbog iste kalcifikacije. [12], [13]
Dijagnostika alkaptonurija
Obično se dijagnoza kongenitalnih metaboličkih poremećaja temelji na proučavanju tjelesnih tekućina.
Na temelju kojih se testova i kakvih reakcija može dijagnosticirati alkaptonurija? Potrebni su testovi urina - za otkrivanje homogentizinske kiseline i određivanje njezine razine (normalna - 20-30 mg dnevno, povećana - 3-8 g). Uzorak urina ispituje se plinskom kromatografijom ili masenom spektrometrijom pomoću tekuće kromatografije; moguć je skrining test na prisutnost željeznog klorida u urinu. [14]
Postoji i brza dijagnostička metoda - određivanje alkaptona u osušenim mrljama urina na papiru (po intenzitetu boje).
Prilikom razjašnjavanja dijagnoze, instrumentalna dijagnostika (RTG) uključuje identifikaciju rentgenskih znakova osteoartritisa i drugih zglobnih patologija u pacijenata.
Dijagnoza se potvrđuje takvim& molekularno genetskim metodama kao što je dijagnostika nasljednih bolesti , poput genetskog testiranja i sekvenciranja DNA. [15]
Diferencijalna dijagnoza
Diferencijalna dijagnoza uključuje hemokromatozu i akutno zatajenje jetre novorođenčadi, melaninuriju, akutnu povremenu porfiriju, hemofagocitnu limfohistiocitozu, primarnu mitohondrijsku patologiju, reumatoidni artritis, ankilozirajući spondilitis.
Tko se može obratiti?
Liječenje alkaptonurija
Glavni način liječenja alkaptonurije je unos velikih doza (najmanje 1000 mg dnevno) askorbinske kiseline. U djece se time povećava izlučivanje homogentizinske kiseline u mokraći, a u odraslih smanjuje razinu njezinog derivata, benzokinoctene kiseline, u urinu i usporava njezino vezivanje za strukture vezivnog tkiva zglobova i kolagen. [16]
U zapadnoeuropskim klinikama testira se lijek Nitizinone (Orfalin), lijek iz skupine metabolita koji inhibira drugu fazu katabolizma tirozina: transformaciju para-hidroksifenilpiruvata u homogentizinsku kiselinu. Međutim, uporaba ovog farmakološkog sredstva dovodi do nakupljanja tirozina i može uzrokovati ozbiljne nuspojave, uključujući zamućenje rožnice i fotofobiju, krvarenje iz nosa i želuca, zatajenje jetre, promjene u krvi itd. Međutim, u Sjedinjenim Državama Nithizinone je odobren od strane FDA za liječenje tirozinemije tipa I. [17], [18]
Kao takva, fizioterapija - terapija vježbama za povećanje mišićne snage i povećanje pokretljivosti zglobova, balneo i peloidoterapija za ograničavanje boli - provodi se za probleme sa zglobovima uzrokovane alkaptonurijom.
Iako se tirozin ne opskrbljuje samo hranom, već se i proizvodi u tijelu, pacijentima s alkaptonurijom preporučuje se prehrana siromašna bjelančevinama i ograničavanje unosa hrane bogate tirozinom, prvenstveno govedine i svinjetine, mliječnih proizvoda (osobito sireva), mahunarki, orasi itd. Sjemenke.
Prevencija
Sprječavanje genskih mutacija je nemoguće, a kako bi se spriječilo rođenje djece s visokim rizikom od urođenih poremećaja, postoji medicinsko i genetsko savjetovanje, koje je potrebno prije planirane trudnoće onih bračnih parova u čijoj obiteljskoj anamnezi postoje bolesti nasljednog karaktera priroda. [19]
Prognoza
U tijeku alkaptonurije vrlo je mali broj smrtnih slučajeva, a smrt može biti uzrokovana ozbiljnim komplikacijama koje pogađaju srce i bubrege. Dakle, za ukupni životni vijek osoba s alkaptonurijom, prognoza je povoljna.
No, kvaliteta života je smanjena zbog intenzivnih bolova u zglobovima ili kralježnici sa značajnim ograničenjem pokretljivosti, često progresivnim.