Medicinski stručnjak članka

Nove publikacije
Angelmanov sindrom kod djece i odraslih
Posljednji pregledao: 04.07.2025

Svi iLive sadržaji medicinski se pregledavaju ili provjeravaju kako bi se osigurala što je moguće točnija činjenica.
Imamo stroge smjernice za pronalaženje izvora i samo povezujemo s uglednim medijskim stranicama, akademskim istraživačkim institucijama i, kad god je to moguće, medicinski pregledanim studijama. Imajte na umu da su brojevi u zagradama ([1], [2], itd.) Poveznice koje se mogu kliknuti na ove studije.
Ako smatrate da je bilo koji od naših sadržaja netočan, zastario ili na neki drugi način upitan, odaberite ga i pritisnite Ctrl + Enter.

Postoji niz bolesti za koje izrazi poput "pazi na sebe i nećeš se razboljeti" zvuče, u najmanju ruku, smiješno. To su patologije kod kojih su neke mentalne i fizičke abnormalnosti svojstvene djetetovom tijelu čak i prije rođenja, ali roditelji nisu krivi za to. Takve bolesti uzrokovane su mutacijama ili abnormalnostima u kromosomskim setovima i nazivaju se kromosomskim ili genetskim. Angelmanov sindrom, Downov sindrom, Patauov sindrom, Edwardsov sindrom, Turnerov sindrom, Prader-Willijev sindrom - ovo je samo dio genetskih bolesti s prilično pristojnog popisa.
Sindrom sretnog čovjeka
Ovaj put ćemo govoriti o patologiji nazvanoj po engleskom pedijatru Harryju Angelmanu, koji je prvi put pokrenuo pitanje ovog problema 1965. godine, susrevši se dan ranije u svojoj praksi s troje neobične djece, ujedinjene zajedničkim osebujnim simptomima. Liječnik je ovu djecu nazvao djecom lutkama i napisao članak o njima, koji se u početku zvao "Djeca-marionete". Sam članak i njegov naslov napisani su pod dojmom slike viđene u jednom od muzeja Verone. Slika je prikazivala dječaka koji se smije, a zvala se "Dječak lutka". Povezanost djeteta prikazanog na slici s troje djece koju je Angelman jednom susreo u svojoj praksi potaknula je pedijatra da djecu spoji u jednu skupinu zbog bolesti koju su imali.
Nema ništa iznenađujuće u činjenici da djecu spomenutu u članku nisu primijetili drugi liječnici. Uostalom, na prvi pogled se činilo da imaju potpuno različite bolesti, toliko je bila različita opća klinička slika bolesti u 3 različita slučaja. Možda bi "nova" kromosomska patologija zainteresirala druge znanstvenike, ali u to vrijeme genetika još nije bila dovoljno razvijena da potvrdi hipotezu engleskog liječnika. Stoga je, nakon određenog interesa za nju, članak dugo bačen na stražnju policu.
Sljedeći spomen Angelmanovog sindroma, kako se sada zvao članak engleskog pedijatra G. Angelmana, datira iz ranih 80-ih godina 20. stoljeća. I tek je 1987. godine bilo moguće pronaći razlog zašto se mali dio djece rađa s takvim odstupanjima da se izvana čini da su stalno nasmijani i sretni. Zapravo, to uopće nije istina, a osmijeh je samo grimasa, iza koje se krije nesretna ljudska duša i bol roditelja.
Epidemiologija
Prema statistikama, kromosomska mutacija kod djeteta može se razviti i na pozadini sličnih mutacija kod roditelja i u odsutnosti takvih. Ne postoji jasna nasljedna priroda Angelmanovog sindroma (AS), ali vjerojatnost razvoja patologije kod roditelja s kromosomskim mutacijama je prilično visoka.
Zanimljivo je i da ako obitelj već ima dijete s AS-om, postoji jedan posto šanse da će imati drugo dijete s istim poremećajem, čak i ako su roditelji zdravi.
Još uvijek ne postoje točne statistike o broju pacijenata s Angelmanovim sindromom. Možda je razlog raznolikost simptoma, koji se mogu pojaviti u određenom sastavu ili se uopće ne pojaviti dulje vrijeme. Pretpostavlja se da je prevalencija bolesti: 1 dijete na 20 000 novorođenčadi. Ali ta je brojka vrlo približna.
Uzroci Angelmanov sindrom
Angelmanov sindrom je medicinski naziv za kromosomsku patologiju, ali daleko od jedinog. Ljudi ovu bolest nazivaju sindromom djece lutaka, sindromom sretne lutke, Petruškinim sindromom i sindromom lutke koja se smije. Ljudi smišljaju svakakve nazive (ponekad čak i uvredljive za same pacijente i njihove roditelje), ali bolest je bolest, bez obzira koliko smiješno izgledala i bez obzira na razloge.
A razlozi za razvoj Angelmanovog sindroma, kao i mnogih drugih genetskih patologija, u svim slučajevima su poremećaji u strukturi jednog od kromosoma ili kromosomskog skupa u cjelini. Ali u našem slučaju, cijeli problem leži u kromosomu 15, prenesenom od majke. To jest, očinski kromosom u ovom slučaju nema odstupanja, ali ženski prolazi kroz određene mutacije.
Prema vrsti kromosomske abnormalnosti, Angelmanov sindrom se klasificira kao kromosomska mutacija. Takve mutacije se smatraju:
- Delecija (odsutnost dijela kromosoma koji sadrži određeni skup gena; ako nedostaje jedan od gena, govorimo o mikrodeleciji), koja je rezultat dva prekida i jednog ponovnog spajanja, kada se izgubi dio izvornog kromosoma.
- Duplikacija (prisutnost dodatnog dijela kromosoma koji je kopija postojećeg), koja u većini slučajeva dovodi do smrti osobe, a rjeđe do neplodnosti.
- Inverzija (zaokret jednog od dijelova kromosoma za 180 stupnjeva, tj. u suprotnom smjeru, a zatim se geni u njemu nalaze suprotnim redoslijedom), kada se prekinuti krajevi kromosoma spajaju redoslijedom drugačijim od izvornog.
- Umetanje (ako dio genetskog materijala u kromosomu nije na svom mjestu),
- translokacija (ako je određeni dio kromosoma pričvršćen na drugi kromosom; takva mutacija može biti obostrana bez gubitka dijelova).
Primajući mutirani kromosom od ništa ne sluteće majke, dijete je osuđeno na rođenje s abnormalnostima. Najčešćim uzrokom Angelmanovog sindroma i dalje se smatra delecija majčinog 15. kromosoma, kada nedostaje mali dio. Manje česte mutacije u sindromu "lutke koja se smije" smatraju se:
- translokacija,
- unipaternalna disomija (ako je dijete primilo par kromosoma od oca, majčin kromosom je odsutan),
- mutacija gena u DNK, koji su i glavni gradivni (genetski) materijal i upute za njegovu ispravnu upotrebu (posebno mutacija gena ube3a u majčinom kromosomu).
Prisutnost jedne od ovih mutacija kod roditelja faktor je rizika za razvoj Angelmanovog sindroma kod djece. Ali ne samo kromosomske mutacije, već i genomske (koje su povezane s kvantitativnom promjenom kromosomskih setova i češće su od kromosomskih) mogu izazvati razvoj bolesti kod djeteta. Uobičajene genomske mutacije uključuju trisomiju kromosoma (ako kromosomski set osobe ima više od 46 kromosoma).
Da bi se patologija pojavila kod djeteta, uopće nije potrebno da roditelji imaju kromosomske abnormalnosti. Ipak, postoji određeni postotak pacijenata čija je bolest nasljedna.
Patogeneza
Zaronimo malo dublje u biologiju, ili preciznije, genetiku. Genetska informacija svakog pojedinog ljudskog organizma sadržana je u 23 para kromosoma. Jedan kromosom iz para prenosi se na dijete od oca, drugi od majke. Svi parovi kromosoma razlikuju se po obliku i veličini te nose određene informacije. Dakle, 23. par kromosoma (X i Y kromosomi) odgovoran je za formiranje spolnih karakteristika bebe (XX - djevojčica, XY - dječak, dok Y kromosom dijete može primiti samo od oca).
Idealno bi bilo da dijete od roditelja naslijedi 46 kromosoma koji formiraju njegove genetske karakteristike, predodređujući ga kao pojedinca. Veći broj kromosoma naziva se trisomija i smatra se odstupanjem od norme. Na primjer, prisutnost kromosoma 47 u kromosomskom skupu (kariotip, koji određuje vrstu i individualne karakteristike) uzrokuje pojavu Downovog sindroma.
Ako se kromosomi oboje posebnom bojom, tada se pod mikroskopom mogu vidjeti pruge različitih nijansi duž svake od njih. Unutar svake pruge nalazi se ogroman broj gena. Sve te pruge znanstvenici su numerirali i imaju fiksnu lokaciju. Odsutnost jedne od pruga smatra se odstupanjem od norme. Kod Angelmanovog sindroma vrlo često se može uočiti odsutnost segmenata majčinog kromosoma u intervalu q11-q13, smještenog u dugom kraku, čiji je broj DNA baza samo oko 4 milijuna.
Glavna komponenta kromosoma smatra se nevjerojatno dugom molekulom DNA koja sadrži tisuće gena i desetke i stotine milijuna dušičnih baza. Dakle, kromosom 15, odgovoran za razvoj Angelmanovog sindroma i nekoliko drugih, sadrži 1200 gena i oko 100 milijuna baza. Bilo kakvi poremećaji u strukturi molekule DNA sigurno će utjecati na izgled i razvoj budućeg djeteta.
Genetska informacija sadržana u genima pretvara se u protein ili RNA. Taj se proces naziva ekspresija gena. Na taj način, genetska informacija primljena od roditelja dobiva i oblik i sadržaj, što je utjelovljeno u njihovom jedinstvenom ženskom ili muškom nasljedniku.
Postoji niz patologija s neklasičnim tipom nasljeđivanja, uključujući Angelmanov sindrom, u kojem geni primljeni od roditelja kao dio uparenih kromosoma nose jedinstveni otisak roditelja i manifestiraju se na različite načine.
Dakle, Angelmanov sindrom je upečatljiv primjer genomskog imprintinga, u kojem je ekspresija gena u djetetovom tijelu izravno ovisna o tome od kojeg su roditelja primljeni aleli (različiti oblici jednog gena, primljeni od oca i majke, smješteni na identičnim dijelovima uparenih kromosoma). To jest, samo anomalije na majčinom kromosomu dovode do razvoja sindroma, dok mutacije i strukturni poremećaji očinskog kromosoma uzrokuju potpuno različite patologije.
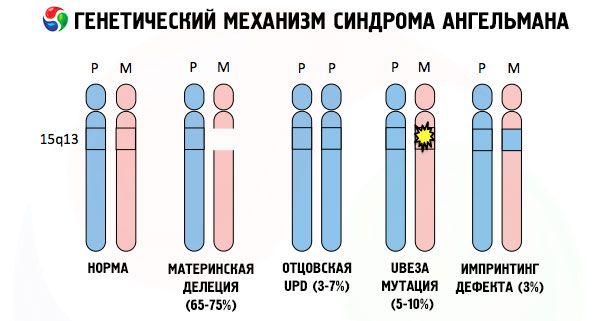
Kod ove patologije postoji nedostatak određenih gena u majčinom kromosomu ili gubitak/smanjenje aktivnosti pojedinačnih gena (u velikoj većini slučajeva gena ube3a, koji je uključen u metabolizam ubikvitina, proteina koji regulira razgradnju drugih proteina). Kao rezultat toga, djetetu se dijagnosticiraju mentalne razvojne abnormalnosti i fizičke deformacije.
Simptomi Angelmanov sindrom
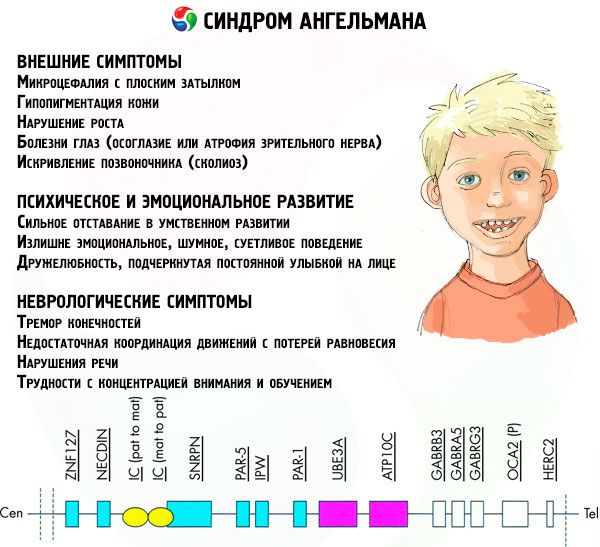
Simptomi Angelmanovog sindroma utječu na različite aspekte djetetovog života i razvoja: fizičke, neurološke, mentalne. Na temelju toga mogu se identificirati 3 skupine simptoma koje ukazuju na razvoj ove patologije.
- Vanjski ili fizički simptomi:
- nesrazmjerno mala glava u usporedbi s tijelom i udovima, koji su normalne veličine,
- preširoka usta,
- gotovo uvijek je osmijeh na licu (s otvorenim ustima),
- rijetki zubi,
- uska gornja usna,
- često isplazivan širok jezik,
- izbočena donja čeljust,
- šiljasta brada,
- vrlo svijetla koža, često kosa (albinizam, povezan s činjenicom da tijelo ne proizvodi pigment melanin),
- tamne mrlje na svijetloj koži (hipopigmentacija zbog nedovoljne proizvodnje melanina)
- fizički ili vanjski simptomi: očne bolesti poput strabizma ili atrofije vidnog živca,
- zakrivljenost kralježnice (skolioza),
- ukočene noge (pri hodanju osoba ne savija noge u koljenima zbog male pokretljivosti zglobova, otuda usporedba s hodom lutke).
- Simptomi povezani s mentalnim i emocionalnim razvojem:
- teška mentalna retardacija,
- pretjerano emocionalno, bučno, nervozno ponašanje,
- često pljeskanje rukama,
- izražena prijateljska naklonost, naglašena stalnim osmijehom na licu,
- česti smijeh bez razloga.
- Neurološki simptomi:
- tremor udova,
- nedovoljna koordinacija pokreta s gubitkom ravnoteže,
- smanjen mišićni tonus,
- razni poremećaji spavanja,
- česti histerični napadaji u djetinjstvu,
- poremećaji govora (dijete kasno počinje govoriti, ima slabe komunikacijske vještine i nerazgovijetan govor),
- hiperaktivnost na pozadini povećane razdražljivosti,
- poteškoće s koncentracijom i učenjem.
Ali ovo je generalizirana slika bolesti. Zapravo, klinička slika Angelmanovog sindroma uvelike ovisi o stadiju razvoja bolesti i vrsti kromosomske mutacije koja je uzrokovala patologiju. To znači da se simptomi bolesti mogu značajno razlikovati kod različitih pacijenata, što nam dugo nije dopuštalo da razlikujemo patologiju od drugih sa sličnom kliničkom slikom.
Među ukupnim brojem simptoma možemo istaknuti one koji su karakteristični za sve pacijente bez iznimke:
- teška mentalna retardacija,
- neprimjereno ponašanje (nerazuman smijeh, povećana razdražljivost, slaba koncentracija, stanje euforije),
- nerazvijenost motoričkih sposobnosti,
- loša koordinacija pokreta, ataksija hoda (neravnomjeran tempo, ljuljanje s jedne strane na drugu itd.), tremor udova.
- poremećaj razvoja govora s prevlasti neverbalnih sredstava komunikacije.
Među simptomima koje susreće velika većina pacijenata mogu se razlikovati sljedeći:
- nesrazmjer između glave i tijela uzrokovan zakašnjelim fizičkim razvojem,
- kod mnogih pacijenata oblik lubanje je takav da veličina mozga ostaje manja nego kod zdravih osoba (mikrocefalija),
- epileptički napadaji prije 3 godine života s progresivnim smanjenjem jačine i učestalosti u starijoj dobi,
- izobličenje EEG parametara (fluktuacije i visoka amplituda niskofrekventnih valova).
Ovi simptomi su prilično česti, međutim, 20% pacijenata s Angelmanovim sindromom ih nema.
Još rjeđe je moguće dijagnosticirati takve manifestacije bolesti kao što su:
- teški ili blagi strabizam,
- loša kontrola pokreta jezika, što rezultira time da pacijenti često isplažuju jezik bez ikakvog razloga,
- poteškoće s gutanjem i sisanjem, posebno kod male djece,
- poremećaj pigmentacije kože i očiju,
- ruke podignute ili savijene pri hodanju,
- hiperrefleksija,
- poremećaji spavanja, posebno u djetinjstvu,
- često slinjenje,
- nezasitna žeđ,
- preaktivni žvakaći pokreti,
- preosjetljivost na toplinu,
- ravni stražnji dio glave,
- izbočena donja čeljust,
- glatki dlanovi.
Prilično velik postotak pacijenata ima problema s mokrenjem, koje slabo kontroliraju, oštećenu finu motoriku, što stvara poteškoće u samobrizi i učenju, te prekomjernu težinu. Gotovo svi pacijenti pubertet doživljavaju kasnije od zdravih vršnjaka.
Djeca s Angelmanovim sindromom dobro percipiraju usmeni govor i razumiju ga, ali ne žele sudjelovati u razgovoru, ograničavajući svoj govor na nekoliko desetaka riječi potrebnih u svakodnevnom životu. Međutim, u odrasloj dobi takvi pacijenti izgledaju mlađe od svojih vršnjaka bez genetskih patologija.
Mnogi simptomi Angelmanovog sindroma su promjenjivi, pa se klinička slika bolesti značajno mijenja s godinama. Konvulzije i epileptički napadaji postaju rjeđi ili potpuno nestaju, pacijent postaje manje uzbuđen, a san se poboljšava.
Komplikacije i posljedice
Angelmanov sindrom je teška, trenutno praktički neizlječiva kromosomska patologija koja pacijentima oduzima mogućnost normalnog života. Kakav će biti život djeteta s AS-om uvelike ovisi o vrsti kromosomske abnormalnosti.
Duplikacija kromosomskog segmenta u većini je slučajeva nespojiva sa životom. Čak i ako takvi pacijenti ne umru u djetinjstvu i ne dosegnu pubertet, nemaju šanse imati djecu.
Brisanje ili odsutnost dijela gena koji se najčešće javlja kod Angelmanovog sindroma predstavlja prepreku djetetu da nauči hodati i govoriti. Takva djeca imaju teži oblik mentalne retardacije, a epileptički napadaji se javljaju češće, a njihov intenzitet je mnogo veći nego kod pacijenata s drugim kromosomskim abnormalnostima.
Ako postoji samo mutacija jednog gena, uz dužnu pažnju i pristup dijete se može naučiti osnovama brige o sebi, komunikacije i interakcije u skupini, iako će i dalje zaostajati za svojim vršnjacima u razvoju.
Za djecu s Angelmanovim sindromom, koja su po prirodi ljubazna, najvažnija je ljubav i pažnja roditelja. Samo u tom slučaju djetetovo obrazovanje će donijeti plodove, čak i ako su mali. Naravno, pacijenti s AS-om neće moći učiti u redovnoj školi. Potrebni su im posebni razredi u kojima će se djeca prvo učiti koncentraciji, a zatim će im se postupno davati osnove školskog znanja.
Dijagnostika Angelmanov sindrom
Angelmanov sindrom je kongenitalna razvojna patologija. No, zbog određenih okolnosti, često ga je nemoguće dijagnosticirati u dojenačkoj dobi i ranom djetinjstvu. To je zbog nespecifičnosti i slabe ekspresije simptoma kod dojenčadi i djece mlađe od 3 godine. A prevalencija bolesti u našoj zemlji nije toliko velika da su je liječnici naučili prepoznati među njezinim vršnjacima.
Angelmanov sindrom kod dojenčadi može se manifestirati kao smanjeni mišićni tonus, što se manifestira problemima s hranjenjem (slabost refleksa sisanja i gutanja), a kasnije i poteškoćama u učenju hodanja (takva djeca počinju hodati mnogo kasnije). Ovi simptomi su prvi znakovi razvojne abnormalnosti kod bebe, koja može biti povezana s kromosomskom abnormalnošću. Samo genetska analiza može potvrditi ovu pretpostavku.
Posebna se pozornost posvećuje djeci čiji roditelji imaju različite genomske ili kromosomske poremećaje. Uostalom, bolest se možda isprva ne manifestira, a ako se patologija otkrije na vrijeme, intenzivnim početkom rada s djetetom moguće je postići znatno veći uspjeh u učenju, usporavajući napredovanje bolesti.
Ako roditelji imaju različite kromosomske abnormalnosti, genetska analiza se provodi čak i prije rođenja djeteta, budući da je SA jedna od patologija koje se mogu otkriti u embrionalnoj fazi.
Prikupljanje materijala za genetska istraživanja može se provesti na dva načina:
- invazivan (s određenim postotkom rizika, budući da je potrebno prodrijeti u maternicu kako bi se uzeo uzorak amnionske tekućine),
- neinvazivna (analiza DNK djeteta iz majčine krvi).
Zatim se provode sljedeće studije:
- fluorescentna in situ hibridizacija (FISH metoda) – vezanje DNA sonde obilježene posebnom bojom na DNA koja se proučava, nakon čega slijedi pregled pod mikroskopom.
- analiza mutacija u genu ube3a i imprintiranim genima,
- Analiza metilacije DNA korištenjem posebnih metoda koje se koriste u genetici.
Genetski testovi pružaju prilično točne informacije u slučaju kromosomskih abnormalnosti, što znači da budući roditelji unaprijed znaju na što se trebaju pripremiti. Međutim, postoje iznimke. U određenoj skupini pacijenata, uz prisutnost svih simptoma koji ukazuju na patologiju, rezultati testova ostaju normalni. To jest, patologija se može prepoznati samo pažljivim promatranjem djeteta od ranog djetinjstva: kako jede, kada je počelo hodati i govoriti, savija li noge pri hodanju itd.
Uz FISH metodu, među instrumentalnim dijagnostičkim metodama za Angelmanov sindrom mogu se izdvojiti tomografija (CT ili MRI), koja pomaže u određivanju stanja i veličine mozga, te elektroencefalogram (EEG), koji pokazuje kako funkcioniraju pojedini dijelovi mozga.
Liječnici obično postavljaju konačnu dijagnozu u dobi od 3-7 godina, kada pacijent već ima većinu simptoma i vidljiva je dinamika razvoja bolesti.
Koji su testovi potrebni?
Diferencijalna dijagnoza
Angelmanov sindrom je genetska patologija koja praktički nema specifičnih manifestacija. Većina simptoma može podjednako ukazivati i na AS i na druge genetske patologije.
Diferencijalna dijagnoza Angelmanovog sindroma provodi se sa sljedećim patologijama:
- Pitt-Hopkinsov sindrom (pacijente karakterizira mentalna retardacija, vedar karakter, nasmijani su, imaju prilično velika i široka usta, primjećuje se mikrocefalija). Razlika je u napadima hiperventilacije i zadržavanja daha u budnom stanju.
- Christiansonov sindrom (pacijenti su mentalno zaostale osobe veselog raspoloženja, nesposobne za govor, karakterizirane mikrocefalijom, ataksijom, konvulzijama, nevoljnim pokretima mišića).
- Mowat-Wilsonov sindrom (simptomi: mentalna retardacija, epileptički napadaji, šiljasta brada, otvorena usta, sretan izraz lica, mikrocefalija). Karakteristike: velika udaljenost između očiju, oči koso prema unutra, zaobljeni vrh nosa, ušna školjka okrenuta unatrag.
- Kabukijev sindrom (karakterizira ga blaga do umjerena mentalna retardacija, problemi s govorom i motorikom, slabost mišića, epileptički napadaji, mikrocefalija, dugi intervali između svrbeža i poremećena koordinacija). Karakteriziraju ga lučne obrve, izvijen lateralni dio donjeg kapka, široko postavljene oči, duge palpebralne pukotine s dugim, gustim trepavicama.
- Rettov sindrom (diferencijacija od AS-a kod žena). Simptomi: usporeni razvoj govora, napadaji, mikrocefalija. Razlika je u tome što nema sretnog izraza lica, javljaju se napadi apneje i apraksije, što s vremenom napreduje.
- Autosomno recesivni sindrom mentalne tardacije 38 (simptomi: izražena mentalna retardacija s kašnjenjima u motoričkim sposobnostima i govoru, slabost mišića, problemi s hranjenjem u dojenačkoj dobi, impulzivnost). Karakteristična značajka je plava boja šarenice.
- Sindrom duplikacije gena MECP 2 (diferencijacija od SA kod muškaraca). Simptomi: teška mentalna retardacija, slabost mišića od djetinjstva, problemi s govorom ili nedostatak govora, epilepsija. Karakteristike: progresivna miopatija, stalno ponavljajuće infekcije.
- Kleefstraov sindrom (simptomi: problemi s govorom i razmišljanjem, slabost mišića, poremećaji spavanja, nedostatak pažnje, otvorena usta, hiperaktivnost, napadaji, ataksija, poremećaji ravnoteže). Karakteristične značajke: ravno lice, kratak prćast nos, široko postavljene oči, velika izvijena donja usna, agresivni ispadi.
- Smith-Magenisov sindrom (karakteriziraju ga napadaji, problemi sa spavanjem, poremećaji intelektualnog i motoričkog razvoja). Karakteristične značajke uključuju široko i ravno lice te istaknuto čelo.
- Koolen-de Vriesov sindrom (blaga do umjerena mentalna retardacija, slabost mišića, napadaji, prijateljski nastrojenost). Karakteristične značajke: dugo lice s visokim čelom, izbočene uši, kose oči, velika pokretljivost zglobova, kongenitalne srčane mane.
- Phelan-McDermidov sindrom (simptomi: mentalna retardacija, poremećaji govora ili nedostatak govora). Karakteristike: velike ruke s razvijenim mišićima, slabost mišića od rođenja, slabo znojenje.
Patologije poput nedostatka adenil sukcinata, sindroma autosomno recesivnog mentalnog zaostajanja 1, sindroma duplikacije kromosoma 2q23.1, sindroma haploinsuficijencije gena FOXG1, STXBP1 ili MEF2C i nekih drugih mogu se "pohvaliti" simptomima sličnim Angelmanovom sindromu.
Zadatak liječnika je postaviti točnu dijagnozu, razlikovati Angelmanov sindrom od patologija sa sličnim simptomima i propisati učinkovito liječenje koje je relevantno za dijagnosticirani stadij bolesti.
Liječenje Angelmanov sindrom
Angelmanov sindrom jedna je od onih patologija za koje medicina još uvijek traži učinkovito liječenje. Etiološko liječenje bolesti je u fazi razvoja različitih metoda i sredstava, od kojih mnoga još nisu testirana na ljudima. To znači da se liječnici zasad moraju ograničiti na simptomatsku terapiju, koja pomaže nekako ublažiti nezavidnu situaciju djece i odraslih s marionetskim sindromom, koji pate od epileptičkih napadaja, salivacije, hipotenzije i poremećaja spavanja.
Dakle, moguće je smanjiti učestalost i jačinu epileptičkih napadaja uz pomoć pravilno odabranog antikonvulzivnog lijeka. Ali cijela poteškoća je u tome što se napadaji kod pacijenata sa SA razlikuju od običnih epileptičkih napadaja po tome što ih karakterizira nekoliko vrsta napadaja, što znači da se stanje može ublažiti primjenom nekoliko lijekova odjednom.
Najpopularniji antikonvulzivi koji se koriste za liječenje Angelmanovog sindroma su: valproična kiselina, topiramat, lamotrigin, levetiracetam, klonazepam i lijekovi na njihovoj bazi. Manje često se koriste lijekovi na bazi karmazepina, fenitoina, fenobarbitala, etosuksimida, budući da neki od njih mogu izazvati paradoksalni učinak koji se sastoji u pojačavanju i povećanju učestalosti epileptičkih napadaja. To se događa ako se lijek koristi kao dio monoterapije.
Za liječenje slinjenja obično se koriste dvije metode: medikamentozna (lijekovi koji potiskuju proizvodnju sline) i kirurška, koja uključuje reimplantaciju slinovnica. No, u slučaju salmonalemije, ove se metode smatraju neučinkovitima i pitanje ostaje otvoreno. Roditelji i oni koji brinu o takvim pacijentima moraju posvetiti posebnu pozornost ovom problemu, budući da sami pacijenti obično ne kontroliraju slinjenje, a neki se jednostavno ne mogu sami brinuti o sebi.
Drugi problem je kratko trajanje sna. Djeca s Angelmanovim sindromom često spavaju najviše 5 sati, što negativno utječe na funkcioniranje cijelog tijela. Lako uzbuđena, aktivna djeca koja vole igre i komunikaciju (čak i ako se pokušavaju ograničiti na neverbalne metode) primjetno su umorna tijekom dana. Da bi se dobro odmorilo, tijelu je potreban dubok, potpun san, ali upravo je u tome kvaka.
Čini se da bi sedativni lijekovi (fenotiazini i atipični antipsihotici) koji smiruju živčani sustav trebali biti dovoljni za poboljšanje sna kod uznemirenih pacijenata. No, u slučaju AS-a, upotreba takvih lijekova puna je pojave negativnih učinaka. Stoga liječnici i dalje preferiraju blage tablete za spavanje, poput melatonina (prirodnog hormonskog lijeka na bazi hormona spavanja), koji se pacijentima daje sat vremena prije spavanja u količini od 1 tablete, i difenhidramina. Učestalost primjene i doziranje određuje liječnik ovisno o stanju i dobi pacijenta.
Ponekad pacijenti s Angelmanovim sindromom imaju problema s probavom i stolicom. Stolicu možete poboljšati laksativima (po mogućnosti biljnim).
Ili možete pristupiti problemu drugačije, kao što su to učinili američki liječnici, na temelju nekih metoda liječenja autizma, jer su mnogi simptomi karakteristični za AS također karakteristični za autizam (impulzivnost, nevoljne kretnje, ponavljajuće radnje, deficit pažnje, problemi u komunikaciji itd.). Primijećeno je da uvođenje hormona sekretina, koji normalizira probavu i stolicu, pozitivno utječe na pažnju pacijenata, a oksitocin pomaže u poboljšanju kognitivnih sposobnosti i pamćenja djeteta te ispravljanju ponašanja.
Istina, sami hormoni nisu dovoljni, posebno kada su u pitanju djeca. Kod Angelmanovog sindroma indicirana je bihevioralna terapija, rad s psihologom i logopedom (poučavanje metodama neverbalne komunikacije i znakovnom jeziku). Obrazovanje takve djece trebalo bi se temeljiti na individualnom programu uz sudjelovanje posebno obučenih učitelja, psihologa i roditelja. Nažalost, to nije svugdje moguće, a obitelji ostaju same sa svojim problemom.
Budući da mnogi mladi pacijenti s AS-om pate od niskog mišićnog tonusa i problema sa zglobovima, velika se pozornost posvećuje fizioterapiji. Najčešće liječnici pribjegavaju primjeni parafinskih aplikacija, elektroforeze i magnetske terapije.
Aktivna tonička masaža i posebne vježbe terapijskog tjelesnog treninga pomoći će bolesnom djetetu da nakon nekog vremena stane na noge i samouvjereno hoda. U tom smislu posebno je korisna akvagimnastika koja se preporučuje kod SA u hladnoj vodi. Povećava tonus mišića i uči dijete da kontrolira svoje tijelo i koordinira pokrete.
Antikonvulzivno liječenje
Najopasniji simptom Angelmanovog sindroma su napadaji slični onima kod epilepsije. Ovaj simptom se opaža kod 80% pacijenata, što znači da svima njima treba propisati učinkovitu antikonvulzivnu terapiju.
Liječenje epileptičkih napadaja provodi se uz pomoć vitamina i antikonvulziva. Kod Angelmanovog sindroma, praćenog konvulzivnim sindromom, korisni su vitamini skupine B, kao i vitamini C, D i E. No, samostalno propisivanje vitaminske terapije u ovom slučaju je vrlo opasno, jer nekontrolirani unos vitamina može smanjiti učinkovitost antiepileptika i izazvati nove, teže i dugotrajnije napadaje.
Odabir antikonvulzivnih lijekova i propisivanje njihove učinkovite doze također bi trebao obaviti liječnik specijalist. On ili ona također odlučuje hoće li jedan lijek biti dovoljan ili će pacijent morati uzimati 2 ili više lijekova dulje vrijeme.
Većini pacijenata liječnici propisuju lijekove valproične kiseline (Valproična kiselina, Depakine, Convulex, Valparin itd.), koji sprječavaju napadaje i poboljšavaju raspoloženje i mentalno stanje pacijenata.
Valproična kiselina dostupna je u obliku tableta, sirupa i otopina za injekcije. Najpopularniji lijek je lijek s produljenim oslobađanjem "Depakine" u tabletama i kao otopina za intravenoznu primjenu. Doziranje lijeka određuje liječnik pojedinačno ovisno o težini, dobi i stanju pacijenta.
Lijek se uzima tijekom obroka 2 do 3 puta dnevno. Prosječna dnevna doza je 20-30 mg na 1 kilogram težine pacijenta, maksimalna je 50 mg/kg dnevno.
Kontraindikacije za uporabu. Ne koristiti u slučaju disfunkcije jetre i gušterače, hemoragijske dijateze, hepatitisa, porfirije i preosjetljivosti na lijek.
Nuspojave uključuju tremor ruku, probavne i stolične poremećaje te promjene tjelesne težine.
"Topiramat" je također lijek izbora za SA. Proizvodi se u obliku tableta i koristi se i kao dio monoterapije i u kombinaciji s drugim lijekovima.
Način primjene i doziranje. Tablete uzimajte oralno, neovisno o unosu hrane. Početna dnevna doza za odrasle je 25-50 mg, za djecu - 0,5-1 mg/kg. Svaki tjedan doza se povećava prema uputama liječnika.
Lijek se ne smije uzimati tijekom trudnoće i dojenja, kao ni u slučaju preosjetljivosti na njegove sastojke. Lijek ima mnogo različitih nuspojava.
Lijekovi koje liječnik može propisati za Angelmanov sindrom: Clomazepam, Rivotril, Lamotrigine, Seizar, Lamictal, Levetiracetam, Keppra, Epiterra itd.
 [ 16 ], [ 17 ], [ 18 ], [ 19 ]
[ 16 ], [ 17 ], [ 18 ], [ 19 ]
Tradicionalna medicina i homeopatija
Tradicionalna medicina, poput homeopatskih pripravaka, naravno je relativno sigurna, ali učinkovitost takvog liječenja Angelmanovog sindroma može se smatrati kontroverznom.
Iako narodno liječenje ipak može pomoći u nekim stvarima. Govorimo o zaustavljanju epileptičkih napadaja. U tom smislu, liječenje biljem može biti prilično učinkovito.
Dobar učinak pruža ljekovita zbirka na bazi božura, sladića i vodene leće (komponente se uzimaju u jednakim količinama). Biljke je potrebno samljeti u brašno. Nakon 2 tjedna od početka uzimanja, možete primijetiti značajno smanjenje učestalosti napadaja.
Dekocija lavande (1 čajna žličica po čaši kipuće vode) također je korisna za grčeve. Smjesa se kuha 5 minuta i infuzira pola sata. Lijek se uzima noću 14 dana.
Vodena (ili alkoholna) infuzija majčine trave smatra se učinkovitom kod epileptičkih napadaja.
Od homeopatskih pripravaka za sprječavanje napadaja kod Angelmanovog sindroma, možete koristiti lijekove na bazi kamilice i majčine trave, Acidum hydrocyanicum, Argentum nitricum, Kalium bromatum, Arsenicum album. Ali treba uzeti u obzir da samo homeopatski liječnik može propisati učinkovite i sigurne doze lijekova u svakom konkretnom slučaju.
Prevencija
Kao što je čitatelj vjerojatno već shvatio, medicina još nije u stanju spriječiti genske mutacije i druge kromosomske abnormalnosti, međutim, kao ni ispraviti situaciju. To se može dogoditi bilo kome, jer se djeca s Angelmanovim sindromom rađaju od zdravih roditelja, a genetika, koja je trenutno jedna od najmanje proučavanih grana medicine, to još ne može objasniti.
Jedino što se može učiniti jest odgovorno pristupiti planiranju trudnoće, na vrijeme se registrirati i obaviti preglede. Ali opet, takva će mjera biti više edukativna nego preventivna, kao i svaki pregled. Ali mladi će roditelji unaprijed znati na što se treba pripremiti, a u slučaju pozitivnog odgovora odlučit će mogu li preuzeti takvu odgovornost kao što je odgoj bolesnog djeteta.
Prognoza
Prognoza za Angelmanov sindrom ovisi o prirodi kromosomske abnormalnosti i pravovremenosti njezina otkrivanja. Najteže su pogođena ona djeca čiji kromosom 15 sadrži "praznine" u genima (delecija). Vjerojatnost da takvi pacijenti hodaju i govore izuzetno je niska. Drugi slučajevi mogu se ispraviti pažljivim pristupom i ljubavlju prema svom djetetu.
Nažalost, takvi pacijenti neće moći postati punopravni članovi društva, unatoč činjenici da su daleko od glupih, razumiju govor i njegovo značenje. Međutim, imat će problema s komunikacijom do kraja života. Pacijente se može učiti znakovnom jeziku od djetinjstva, ali ih se ne može prisiliti da komuniciraju riječima. Vokabular "govornih" pacijenata ograničen je na minimum riječi koje se koriste u svakodnevnom životu (5-15 riječi).
Što se tiče očekivanog životnog vijeka i općeg zdravstvenog stanja pacijenata s Angelmanovim sindromom, ovdje se brojke kreću oko prosječnih vrijednosti. U odrasloj dobi pacijenti se uglavnom suočavaju sa zdravstvenim problemima poput skolioze i pretilosti, koji uz pravilan pristup liječenju nisu opasni po život.