Medicinski stručnjak članka

Nove publikacije
Nasljedni nefritis (Alportov sindrom) kod djece
Posljednji pregledao: 05.07.2025

Svi iLive sadržaji medicinski se pregledavaju ili provjeravaju kako bi se osigurala što je moguće točnija činjenica.
Imamo stroge smjernice za pronalaženje izvora i samo povezujemo s uglednim medijskim stranicama, akademskim istraživačkim institucijama i, kad god je to moguće, medicinski pregledanim studijama. Imajte na umu da su brojevi u zagradama ([1], [2], itd.) Poveznice koje se mogu kliknuti na ove studije.
Ako smatrate da je bilo koji od naših sadržaja netočan, zastario ili na neki drugi način upitan, odaberite ga i pritisnite Ctrl + Enter.
Nasljedni nefritis (Alportov sindrom) je genetski određena nasljedna neimuna glomerulopatija, koja se manifestira hematurijom (ponekad s proteinurijom), progresivnim padom bubrežne funkcije s razvojem kroničnog zatajenja bubrega, često u kombinaciji sa senzorineuralnom gluhoćom i oštećenjem vida.
Bolest je prvi put opisao 1902. godine LG Guthrie, koji je promatrao obitelj u kojoj je hematurija uočena u nekoliko generacija. Godine 1915. AF Hurst opisao je razvoj uremije kod članova iste obitelji. Godine 1927. A. Alport prvi je identificirao gubitak sluha kod nekoliko rođaka s hematurijom. Pedesetih godina 20. stoljeća opisane su očne lezije kod slične bolesti. Godine 1972. kod pacijenata s nasljednom hematurijom, tijekom morfološke studije bubrežnog tkiva, Hinglais i sur. otkrili su neravnomjerno širenje i stratifikaciju glomerularnih bazalnih membrana. Godine 1985. identificirana je genetska osnova nasljednog nefritisa - mutacija u genu za kolagen tipa IV (Fiengold i sur., 1985.).
Proučavanje genetske prirode bolesti omogućilo nam je zaključak da su razlike u fenotipskim manifestacijama nasljednog nefritisa (sa ili bez gubitka sluha) posljedica stupnja ekspresije mutiranog gena. Stoga se trenutno sve kliničke varijante smatraju manifestacijama jedne bolesti, a pojam "nasljedni nefritis" sinonim je za pojam "Alportov sindrom".
Prema epidemiološkim studijama, nasljedni nefritis javlja se s učestalošću od 17 na 100 000 djece.
 [
[ Uzroci Alportovog sindroma
Genetska osnova bolesti je mutacija u genu a-5 lanca kolagena tipa IV. Ovaj tip je univerzalan za bazalne membrane bubrega, kohlearni aparat, lećnu kapsulu, mrežnicu i rožnicu oka, što je dokazano u studijama koje koriste monoklonska antitijela protiv ove frakcije kolagena. Nedavno je naznačena mogućnost korištenja DNA sondi za prenatalnu dijagnostiku nasljednog nefritisa.
Naglašava se važnost testiranja svih članova obitelji DNA sondama kako bi se identificirali nositelji mutiranog gena, što je od velike važnosti u provođenju medicinskog i genetskog savjetovanja obitelji s ovom bolešću. Međutim, do 20% obitelji nema rođake koji boluju od bolesti bubrega, što ukazuje na visoku učestalost spontanih mutacija abnormalnog gena. Većina pacijenata s nasljednim nefritisom ima u obitelji osobe s bolestima bubrega, gubitkom sluha i patologijom vida; važni su srodni brakovi između osoba s jednim ili više predaka, budući da se u braku srodnih osoba povećava vjerojatnost primanja istih gena od oba roditelja. Utvrđeni su autosomno dominantni, autosomno recesivni i dominantni, X-vezani putevi prijenosa.
Kod djece se najčešće razlikuju tri vrste nasljednog nefritisa: Alportov sindrom, nasljedni nefritis bez gubitka sluha i familijarna benigna hematurija.
Alportov sindrom je nasljedni nefritis s oštećenjem sluha. Temelji se na kombiniranom defektu u strukturi kolagena glomerularne bazalne membrane bubrega, struktura uha i oka. Gen klasičnog Alportovog sindroma nalazi se u lokusu 21-22 q dugog kraka X kromosoma. U većini slučajeva nasljeđuje se dominantno, vezan za X kromosom. U tom smislu, Alportov sindrom je teži kod muškaraca, budući da je kod žena funkcija mutiranog gena kompenzirana zdravim alelom drugog, neoštećenog kromosoma.
Genetska osnova za razvoj nasljednog nefritisa su mutacije u genima alfa lanaca kolagena tipa IV. Poznato je šest alfa lanaca kolagena tipa IV G: geni a5- i a6-lanaca (Col4A5 i Col4A5) nalaze se na dugom kraku X kromosoma u zoni 21-22q; geni a3- i a4-lanaca (Col4A3 i Col4A4) nalaze se na 2. kromosomu; geni a1- i a2-lanaca (Col4A1 i Col4A2) nalaze se na 13. kromosomu.
U većini slučajeva (80-85%) otkriva se X-vezani obrazac nasljeđivanja bolesti, povezan s oštećenjem gena Col4A5 kao posljedica delecije, točkastih mutacija ili poremećaja splajsiranja. Trenutno je pronađeno više od 200 mutacija gena Col4A5, odgovornih za poremećaj sinteze a5-lanaca kolagena tipa IV. Kod ovog tipa nasljeđivanja bolest se manifestira kod djece oba spola, ali kod dječaka je teža.
Mutacije u lokusima gena Col4A3 i Col4A4 odgovornih za sintezu lanaca a3 i a4 kolagena tipa IV nasljeđuju se autosomno. Prema istraživanjima, autosomno dominantni tip nasljeđivanja opažen je u 16% slučajeva nasljednog nefritisa, a autosomno recesivni tip uočen je u 6% pacijenata. Poznato je oko 10 varijanti mutacija gena Col4A3 i Col4A4.
Rezultat mutacija je kršenje procesa sastavljanja kolagena tipa IV, što dovodi do kršenja njegove strukture. Kolagen tipa IV jedna je od glavnih komponenti glomerularne bazalne membrane, kohlearnog aparata i leće oka, čija će se patologija otkriti u klinici nasljednog nefritisa.
Kolagen tipa IV, koji je dio glomerularne bazalne membrane, sastoji se uglavnom od dva a1-lanca (IV) i jednog a2-lanca (IV), a sadrži i a3, a4, a5-lance. Najčešće, kod X-vezanog nasljeđivanja, mutaciju gena Col4A5 prati odsutnost a3-, a4-, a5- i a6-lanaca u strukturi kolagena tipa IV, a broj o1- i a2-lanaca u glomerularnoj bazalnoj membrani se povećava. Mehanizam ove pojave nije jasan, pretpostavlja se da su uzrok posttranskripcijske promjene u mRNA.
Odsutnost lanaca a3, a4 i a5 u strukturi kolagena tipa IV glomerularnih bazalnih membrana dovodi do njihovog stanjivanja i krhkosti u ranim fazama Alportovog sindroma, što se klinički češće manifestira hematurijom (rjeđe hematurijom s proteinurijom ili samo proteinurijom), gubitkom sluha i lentikonusom. Daljnje napredovanje bolesti dovodi do zadebljanja i oštećene propusnosti bazalnih membrana u kasnim fazama bolesti, s proliferacijom kolagena tipa V i VI u njima, što se očituje povećanjem proteinurije i smanjenjem bubrežne funkcije.
Priroda mutacije koja je u osnovi nasljednog nefritisa uvelike određuje njegovu fenotipsku manifestaciju. U slučaju delecije X kromosoma s istovremenom mutacijom gena Col4A5 i Col4A6 odgovornih za sintezu a5- i a6-lanaca kolagena tipa IV, Alportov sindrom kombinira se s leiomiomatozom jednjaka i genitalija. Prema istraživačkim podacima, u slučaju mutacije gena Col4A5 povezane s delecijom, primjećuje se veća težina patološkog procesa, kombinacija oštećenja bubrega s ekstrarenalnim manifestacijama i ranim razvojem kroničnog zatajenja bubrega, u usporedbi s točkastom mutacijom ovog gena.
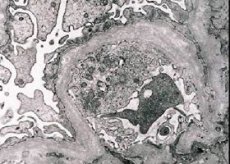
Morfološki, elektronska mikroskopija otkriva stanjivanje i stratifikaciju glomerularnih bazalnih membrana (osobito lamina densa) i prisutnost elektron-denznih granula. Glomerularne lezije mogu biti heterogene kod istog pacijenta, od minimalnih fokalnih mezangijskih lezija do glomeruloskleroze. Glomerulitis kod Alportovog sindroma uvijek je imunonegativan, što ga razlikuje od glomerulonefritisa. Karakteristične značajke uključuju razvoj tubularne atrofije, limfohistiocitičnu infiltraciju i prisutnost "pjenastih stanica" s lipidnim inkluzijama - lipofaga. Kako bolest napreduje, otkriva se zadebljanje i izraženo uništavanje glomerularnih bazalnih membrana.
Otkrivene su određene promjene u imunološkom sustavu. Pacijenti s nasljednim nefritisom imaju smanjenu razinu Ig A i sklonost povećanju koncentracije IgM u krvi, razina IgG može biti povećana u ranim fazama bolesti, a smanjena u kasnijim fazama. Možda je povećanje koncentracije IgM i G vrsta kompenzacijske reakcije kao odgovor na nedostatak IgA.
Funkcionalna aktivnost T-limfocitnog sustava je smanjena; primjećuje se selektivno smanjenje B-limfocita odgovornih za sintezu Ig A, fagocitna veza imuniteta je poremećena, uglavnom zbog poremećaja kemotaksije i unutarstaničnih procesa probave u neutrofilima
Prilikom pregleda biopsije bubrega u bolesnika s Alportovim sindromom, podaci elektronske mikroskopije otkrivaju ultrastrukturne promjene u glomerularnoj bazalnoj membrani: stanjivanje, poremećaj strukture i cijepanje glomerularnih bazalnih membrana s promjenom njezine debljine i neravnim konturama. U ranim fazama nasljednog nefritisa, defekt određuje stanjivanje i krhkost glomerularnih bazalnih membrana.
Stanjivanje glomerularnih membrana je povoljniji znak i češći je kod djevojčica. Konstantniji elektronsko-mikroskopski znak kod nasljednog nefritisa je cijepanje bazalne membrane, a težina njezina uništenja korelira s težinom procesa.
Simptomi Alportovog sindroma kod djece
Prvi simptomi Alportovog sindroma u obliku izoliranog urinarnog sindroma najčešće se otkrivaju kod djece prve tri godine života. U većini slučajeva bolest se otkriva slučajno. Urinarni sindrom se otkriva tijekom preventivnog pregleda djeteta, prije prijema u ustanovu za skrb o djeci ili tijekom ARVI-ja. U slučaju patologije u mokraći tijekom ARVI-ja. Kod nasljednog nefritisa, za razliku od stečenog glomerulonefritisa, nema latentnog razdoblja.
U početnoj fazi bolesti, zdravlje djeteta malo pati, karakteristična značajka je perzistentnost i otpornost urinarnog sindroma. Jedan od glavnih znakova je hematurija različitog stupnja težine, koja se opaža u 100% slučajeva. Povećanje stupnja hematurije primjećuje se tijekom ili nakon respiratornih infekcija, tjelesne aktivnosti ili nakon preventivnih cijepljenja. Proteinurija u većini slučajeva ne prelazi 1 g / dan, na početku bolesti može biti nepostojana, kako proces napreduje, proteinurija se povećava. Povremeno se u urinarnom sedimentu može pojaviti leukociturija s prevlasti limfocita, što je povezano s razvojem intersticijskih promjena.
Naknadno dolazi do djelomičnog oštećenja bubrežne funkcije, pogoršava se opće stanje pacijenta: javljaju se intoksikacija, slabost mišića, arterijska hipotenzija, često se javlja oštećenje sluha (osobito kod dječaka), a ponekad i oštećenje vida. Intoksikacija se manifestira bljedilom, umorom i glavoboljama. U početnoj fazi bolesti, gubitak sluha se u većini slučajeva otkriva samo audiografijom. Gubitak sluha kod Alportovog sindroma može se pojaviti u različitim razdobljima djetinjstva, ali najčešće se gubitak sluha dijagnosticira u dobi od 6-10 godina. Gubitak sluha kod djece počinje visokim frekvencijama, dosežući značajan stupanj u zračnoj i koštanoj vodljivosti, prelazeći iz zvukovodnog u zvukoosjetljivi gubitak sluha. Gubitak sluha može biti jedan od prvih simptoma bolesti i može prethoditi urinarnom sindromu.
U 20% slučajeva, pacijenti s Alportovim sindromom imaju promjene na vidnim organima. Najčešće otkrivene anomalije su one leće: sferofokija, prednji, stražnji ili miješani lentikonus i različite katarakte. U obiteljima s Alportovim sindromom postoji značajna učestalost kratkovidnosti. Brojni istraživači stalno primjećuju bilateralne perimakularne promjene u tim obiteljima u obliku svijetlih bjelkastih ili žućkastih granulacija u žutom tijelu. Smatraju da je ovaj znak stalni simptom koji ima visoku dijagnostičku vrijednost kod Alportovog sindroma. KS Chugh i sur. (1993.) u oftalmološkoj studiji pronašli su kod pacijenata s Alportovim sindromom smanjenje vidne oštrine u 66,7% slučajeva, prednji lentikonus u 37,8%, retinalne pjege u 22,2%, kataraktu u 20% i keratokonus u 6,7%.
Kod neke djece s nasljednim nefritisom, posebno kada se razvije zatajenje bubrega, primjećuje se značajno zaostajanje u fizičkom razvoju. Kako zatajenje bubrega napreduje, razvija se arterijska hipertenzija. Kod djece se češće otkriva u adolescenciji i u starijim dobnim skupinama.
Pacijenti s nasljednim nefritisom karakterizirani su prisutnošću različitih (više od 5-7) stigmi vezivnog tkiva. Među stigmama vezivnog tkiva kod pacijenata najčešći su hipertelorizam očiju, visoko nepce, anomalije zagriza, abnormalan oblik ušnih školjki, zakrivljenost malog prsta na rukama i "sandalni razmak" na stopalima. Nasljedni nefritis karakterizira ujednačenost stigmi dismorfogeneze unutar obitelji, kao i visoka učestalost njihove rasprostranjenosti među rođacima probanda duž čije se linije bolest prenosi.
U ranim stadijima bolesti otkriva se izolirano smanjenje parcijalnih bubrežnih funkcija: transporta aminokiselina, elektrolita, koncentracije, acidogeneze, kasnije promjene utječu na funkcionalno stanje i proksimalnog i distalnog dijela nefrona te su karakterizirane kombiniranim parcijalnim poremećajima. Smanjenje glomerularne filtracije javlja se kasnije, češće u adolescenciji. Kako nasljedni nefritis napreduje, razvija se anemija.
Dakle, nasljedni nefritis karakterizira stadijni tijek bolesti: prvo se javlja latentni stadij ili skriveni klinički simptomi, koji se manifestiraju minimalnim promjenama u urinarnom sindromu, zatim dolazi do postupne dekompenzacije procesa sa smanjenjem bubrežne funkcije s manifestnim kliničkim simptomima (intoksikacija, astenija, zaostajanje u razvoju, anemija). Klinički simptomi se obično pojavljuju bez obzira na slojevitost upalne reakcije.
Nasljedni nefritis može se manifestirati u različitim dobnim razdobljima, što ovisi o djelovanju gena koji je u potisnutom stanju do određenog vremena.
Klasifikacija
Postoje tri vrste nasljednog nefritisa
- Opcija I - klinički se manifestira kao nefritis s hematurijom, gubitkom sluha i oštećenjem oka. Tijek nefritisa je progresivan s razvojem kroničnog zatajenja bubrega. Tip nasljeđivanja je dominantan, vezan za X kromosom. Morfološki se otkriva kršenje strukture bazalne membrane, njezino stanjivanje i cijepanje.
- Opcija II - klinički se manifestira kao nefritis s hematurijom bez gubitka sluha. Tijek nefritisa je progresivan s razvojem kroničnog zatajenja bubrega. Tip nasljeđivanja je dominantan, vezan za X kromosom. Morfološki se otkriva stanjivanje bazalne membrane glomerularnih kapilara (osobito laminadensa).
- Opcija III - benigna familijarna hematurija. Tijek je povoljan, kronično zatajenje bubrega se ne razvija. Vrsta nasljeđivanja je autosomno dominantna ili autosomno recesivna. Kod autosomno recesivnog tipa nasljeđivanja, kod žena se primjećuje teži tijek bolesti.
Dijagnoza Alportovog sindroma
Predlažu se sljedeći kriteriji:
- prisutnost najmanje dva pacijenta s nefropatijom u svakoj obitelji;
- hematurija kao vodeći simptom nefropatije u probanda;
- prisutnost gubitka sluha kod barem jednog člana obitelji;
- razvoj kroničnog zatajenja bubrega kod jednog ili više rođaka.
U dijagnostici različitih nasljednih i kongenitalnih bolesti veliko mjesto se posvećuje sveobuhvatnom pristupu pregledu i, prije svega, obraćanju pažnje na podatke dobivene prilikom sastavljanja djetetovog rodovnika. Dijagnoza Alportovog sindroma smatra se valjanom u slučajevima kada se kod pacijenta otkriju 3 od 4 tipična znaka: prisutnost hematurije i kroničnog zatajenja bubrega u obitelji, prisutnost neurosenzornog gubitka sluha, patologija vida kod pacijenta, otkrivanje znakova cijepanja glomerularne bazalne membrane s promjenom njezine debljine i neravnim konturama tijekom elektronsko-mikroskopskih karakteristika biopsije.
Pregled pacijenta treba uključivati kliničke i genetske metode istraživanja; ciljano proučavanje anamneze bolesti; opći pregled pacijenta uzimajući u obzir dijagnostički značajne kriterije. U fazi kompenzacije, patologija se može otkriti samo fokusiranjem na sindrome kao što su prisutnost nasljednog tereta, hipotenzija, višestruke stigme disembriogeneze, promjene u urinarnom sindromu. U fazi dekompenzacije mogu se pojaviti ekstrarenalni simptomi, poput teške intoksikacije, astenije, usporenog fizičkog razvoja, anemije, koji se manifestiraju i pojačavaju postupnim smanjenjem bubrežne funkcije. Kod većine pacijenata, sa smanjenjem bubrežne funkcije, opaža se sljedeće: smanjena acido- i aminogeneza; 50% pacijenata primjećuje značajno smanjenje sekretorne funkcije bubrega; ograničen raspon fluktuacija optičke gustoće urina; poremećaj filtracijskog ritma, a zatim smanjenje glomerularne filtracije. Stadij kroničnog zatajenja bubrega dijagnosticira se kada pacijenti imaju povišenu razinu uree u krvnom serumu (više od 0,35 g/l) tijekom 3-6 mjeseci ili više, te smanjenje glomerularne filtracije na 25% norme.
Diferencijalnu dijagnozu nasljednog nefritisa treba provoditi prvenstveno s hematurijskim oblikom stečenog glomerulonefritisa. Stečeni glomerulonefritis najčešće ima akutni početak, razdoblje od 2-3 tjedna nakon infekcije, ekstrarenalne znakove, uključujući hipertenziju od prvih dana (kod nasljednog nefritisa, naprotiv, hipotenzija), smanjenu glomerularnu filtraciju na početku bolesti, bez oštećenja parcijalnih tubularnih funkcija, dok su kod nasljednog one prisutne. Stečeni glomerulonefritis javlja se s izraženijom hematurijom i proteinurijom, s povećanom sedimentacijom glomerula. Tipične promjene u bazalnoj membrani glomerula, karakteristične za nasljedni nefritis, imaju dijagnostičku vrijednost.
Diferencijalna dijagnostika od dismetaboličke nefropatije provodi se s kroničnim zatajenjem bubrega, u obitelji klinički otkrivenim heterogenim bolestima bubrega, a može postojati spektar nefropatije od pijelonefritisa do urolitijaze. Djeca se često žale na bol u trbuhu i periodično tijekom mokrenja, u sedimentu urina - oksalate.
Ako se sumnja na nasljedni nefritis, pacijenta treba uputiti na specijalizirani nefrološki odjel radi razjašnjenja dijagnoze.
Što treba ispitati?
Kako ispitati?
Koji su testovi potrebni?
Tko se može obratiti?
Liječenje Alportovog sindroma
Režim uključuje ograničenja teškog fizičkog napora i boravka na svježem zraku. Prehrana je potpuna, s dovoljnom razinom kompletnih proteina, masti i ugljikohidrata, uzimajući u obzir funkciju bubrega. Od velike je važnosti otkrivanje i liječenje kroničnih žarišta infekcije. Koriste se sljedeći lijekovi: ATP, kokarboksilaza, piridoksin (do 50 mg/dan), karnitin klorid. Tečajevi se provode 2-3 puta godišnje. Za hematuriju propisuje se biljni lijek - kopriva, sok od aronije, stolisnik.
U stranoj i domaćoj literaturi postoje izvješća o liječenju prednizolonom i primjeni citostatika. Međutim, teško je procijeniti učinak.
Kod kroničnog zatajenja bubrega koriste se hemodijaliza i transplantacija bubrega.
Ne postoje metode specifične (učinkovite patogenetske) terapije za nasljedni nefritis. Sve mjere liječenja usmjerene su na sprječavanje i usporavanje pada bubrežne funkcije.
Prehrana treba biti uravnotežena i visokokalorična, uzimajući u obzir funkcionalno stanje bubrega. U odsutnosti funkcionalnih poremećaja, prehrana djeteta treba sadržavati dovoljno proteina, masti i ugljikohidrata. U prisutnosti znakova bubrežne disfunkcije, količinu proteina, ugljikohidrata, kalcija i fosfora treba ograničiti, što odgađa razvoj kroničnog zatajenja bubrega.
Tjelesnu aktivnost treba ograničiti; djeci se savjetuje da izbjegavaju sport.
Treba izbjegavati kontakt sa zaraznim pacijentima, smanjiti rizik od razvoja akutnih respiratornih bolesti. Sanacija žarišta kronične infekcije je neophodna. Preventivna cijepljenja se ne provode za djecu s nasljednim nefritisom, cijepljenje je moguće samo zbog epidemioloških indikacija.
Hormonska i imunosupresivna terapija kod nasljednog nefritisa je neučinkovita. Postoje naznake određenog pozitivnog učinka (smanjenje proteinurije i usporavanje progresije bolesti) kod dugotrajne višegodišnje primjene ciklosporina A i ACE inhibitora.
U liječenju pacijenata koriste se lijekovi koji poboljšavaju metabolizam:
- piridoksin - 2-3 mg/kg/dan u 3 doze tijekom 4 tjedna;
- kokarboksilaza - 50 mg intramuskularno svaki drugi dan, ukupno 10-15 injekcija;
- ATP - 1 ml intramuskularno svaki drugi dan, 10-15 injekcija;
- vitamin A - 1000 IU/god./dan u 1 dozi tijekom 2 tjedna;
- Vitamin E - 1 mg/kg/dan u 1 dozi tijekom 2 tjedna.
Ova vrsta terapije pomaže poboljšati opće stanje pacijenata, smanjiti tubularne disfunkcije i provodi se u tečajevima 3 puta godišnje.
Levamisol se može koristiti kao imunomodulator - 2 mg/kg/dan 2-3 puta tjedno s pauzama između doza od 3-4 dana.
Prema istraživačkim podacima, hiperbarična oksigenacija ima pozitivan učinak na težinu hematurije i bubrežne disfunkcije.
Najučinkovitija metoda liječenja nasljednog nefritisa je pravovremena transplantacija bubrega. U tom slučaju nema recidiva bolesti u transplantatu; u malom postotku slučajeva (oko 5%), nefritis se može razviti u transplantiranom bubregu povezan s antigenima na bazalnu membranu glomerula.
Obećavajući smjer je prenatalna dijagnostika i terapija genetskim inženjeringom. Pokusi na životinjama pokazuju visoku učinkovitost prijenosa normalnih gena odgovornih za sintezu alfa lanaca kolagena tipa IV u bubrežno tkivo, nakon čega se opaža sinteza normalnih kolagenih struktura.
Prognoza
Prognoza za nasljedni nefritis je uvijek ozbiljna.
Prognostički nepovoljni kriteriji za tijek nasljednog nefritisa su:
- muški spol;
- rani razvoj kroničnog zatajenja bubrega kod članova obitelji;
- proteinurija (više od 1 g/dan);
- zadebljanje glomerularnih bazalnih membrana prema mikroskopiji;
- akustični neuritis;
- delecija u genu Col4A5.
Prognoza za benignu familijarnu hematuriju je povoljnija.
Использованная литература