Medicinski stručnjak članka

Nove publikacije
Charcot-Marie-Toothova bolest.
Posljednji pregledao: 12.07.2025

Svi iLive sadržaji medicinski se pregledavaju ili provjeravaju kako bi se osigurala što je moguće točnija činjenica.
Imamo stroge smjernice za pronalaženje izvora i samo povezujemo s uglednim medijskim stranicama, akademskim istraživačkim institucijama i, kad god je to moguće, medicinski pregledanim studijama. Imajte na umu da su brojevi u zagradama ([1], [2], itd.) Poveznice koje se mogu kliknuti na ove studije.
Ako smatrate da je bilo koji od naših sadržaja netočan, zastario ili na neki drugi način upitan, odaberite ga i pritisnite Ctrl + Enter.
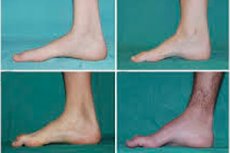
Peronealna mišićna atrofija, Charcot-Marie-Toothov sindrom ili bolest je skupina kroničnih nasljednih bolesti s oštećenjem perifernih živaca.
Prema ICD-10, u odjeljku o bolestima živčanog sustava, šifra za ovu bolest je G60.0 (nasljedna motorna i senzorna neuropatija). Također je uvrštena na popis rijetkih bolesti.
Epidemiologija
Prema kliničkoj statistici, prevalencija svih vrsta Charcot-Marie-Toothove bolesti na 100 tisuća stanovnika iznosi 19 slučajeva (prema drugim izvorima, jedan slučaj na 2,5-10 tisuća stanovnika).
CMT tip 1 čini oko dvije trećine slučajeva (jedan slučaj na 5000 do 7000 stanovnika), a gotovo 70% njih povezano je s duplikacijom gena PMP22. Više od 1,2 milijuna ljudi diljem svijeta pati od ove vrste bolesti.
Incidencija CMT tipa 4 procjenjuje se na 1-5 slučajeva na 10 000 djece. [ 1 ]
Uzroci Charcot-Marie-Toothova bolest
Prema klasifikaciji polineuropatskih sindroma, peronealna (fibularna) mišićna atrofija, Charcot-Marie-Toothova neuralska amiotrofija ili Charcot-Marie-Toothova bolest (skraćeno CMT) odnosi se na genetski određene motorno-senzorne polineuropatije. [ 2 ]
To jest, uzroci njegove pojave su genetske mutacije. I ovisno o prirodi genetskih odstupanja, razlikuju se glavne vrste ili vrste ovog sindroma: demijelinizirajući i aksonski. Prva skupina uključuje Charcot-Marie-Tooth bolest tipa 1 (CMT1), koja nastaje zbog dupliciranja gena PMP22 na kromosomu 17, koji kodira transmembranski periferni mijelinski protein 22. Kao rezultat toga, dolazi do segmentalne demijelinizacije aksonske ovojnice (nastavci živčanih stanica) i smanjenja brzine provođenja živčanog signala. Osim toga, mutacije mogu biti u nekim drugim genima.
Aksonski oblik je Charcot-Marie-Toothova bolest tipa 2 (CMT2), koja zahvaća same aksone i povezana je s patološkim promjenama u genu MFN2 na lokusu 1p36.22, koji kodira membranski protein mitofuzin-2, koji je neophodan za fuziju mitohondrija i stvaranje funkcionalnih mitohondrijskih mreža unutar perifernih živčanih stanica. Postoji više od desetak podtipova CMT2 (s mutacijama u specifičnim genima).
Treba napomenuti da je trenutno identificirano više od stotinu gena čija oštećenja, prenesena nasljeđivanjem, uzrokuju različite podtipove Charcot-Marie-Toothove bolesti. Na primjer, mutacije u genu RAB7 dovode do CMT tipa 2B; promjena gena SH3TC2 (koji kodira jedan od membranskih proteina Schwannovih stanica) uzrokuje CMT tip 4C, koji se manifestira u djetinjstvu i karakterizira ga demijelinizacija motornih i senzornih neurona (postoji desetak i pol oblika tipa 4 ove bolesti).
Rijedak CMT tip 3 (nazvan Dejerine-Sottasov sindrom) počinje se razvijati u ranom djetinjstvu, a uzrokovan je mutacijama u genima PMP22, MPZ, EGR2 i drugim.
Kada se CMT tip 5 pojavi u dobi od 5-12 godina, ne opaža se samo motorna neuropatija (u obliku spastične parapareze donjih ekstremiteta), već i oštećenje optičkog i slušnog živca.
Slabost mišića i optička atrofija (s gubitkom vida), kao i problemi s ravnotežom, tipični su za CMT tip 6. A kod Charcot-Marie-Toothove bolesti tipa 7 ne postoji samo motorno-senzorna neuropatija, već i bolest mrežnice u obliku retinitis pigmentose.
Češća među muškarcima, X-vezana CMT ili Charcot-Marie-Toothova bolest s tetraparezom udova (slabost pokreta i ruku i nogu) je demijelinizirajućeg tipa i smatra se da je rezultat mutacije u genu GJB1 na dugom kraku X kromosoma, koji kodira koneksin 32, transmembranski protein Schwannovih stanica i oligodendrocita koji regulira prijenos živčanih signala. [ 3 ]
Faktori rizika
Glavni faktor rizika za CMT je obiteljska anamneza bolesti, odnosno kod bliskih srodnika.
Prema genetičarima, ako su oba roditelja nositelji autosomno recesivnog gena za Charcot-Marie-Toothovu bolest, rizik od rađanja djeteta koje će razviti ovu bolest iznosi 25%. A rizik da će dijete biti nositelj ovog gena (ali neće imati nikakve simptome) procjenjuje se na 50%.
U slučaju X-vezanog nasljeđivanja (kada se mutirani gen nalazi na X kromosomu žene), postoji 50%-tni rizik da će majka prenijeti gen na sina, koji će razviti CMT. Bolest se možda neće pojaviti kada se rodi žensko dijete, ali kćerini sinovi (unuci) mogu naslijediti defektni gen i bolest će se razviti.
Patogeneza
Kod bilo koje vrste Charcot-Marie-Toothove bolesti, njezina patogeneza je uzrokovana nasljednom anomalijom perifernih živaca: motoričkih (pokretnih) i senzornih (osjetljivih).
Ako je CMT tip demijelinizirajući, tada uništenje ili defekt mijelinske ovojnice koja štiti aksone perifernih živaca dovodi do usporavanja prijenosa živčanih impulsa u perifernom živčanom sustavu - između mozga, mišića i osjetilnih organa.
Kod aksonskog tipa bolesti zahvaćeni su sami aksoni, što negativno utječe na snagu živčanih signala, koja je nedovoljna za potpunu stimulaciju mišića i osjetilnih organa.
Također pročitajte:
Kako se prenosi Charcot-Marie-Toothov sindrom? Defektni geni mogu se nasljeđivati autosomno dominantno ili autosomno recesivno.
Najčešći tip nasljeđivanja, autosomno dominantno, javlja se kada postoji jedna kopija mutiranog gena (koju nosi jedan od roditelja). Vjerojatnost prenošenja CMT-a na svako rođeno dijete procjenjuje se na 50%. [ 4 ]
Kod autosomno recesivnog nasljeđivanja, za razvoj bolesti potrebne su dvije kopije defektnog gena (po jedna od svakog roditelja koji ne pokazuje nikakve znakove bolesti).
U 40-50% slučajeva javlja se autosomno dominantna nasljedna demijelinizacija, tj. CMT tip 1; u 12-26% slučajeva aksonska CMT, tj. tip 2. A u 10-15% slučajeva opaža se X-vezano nasljeđivanje. [ 5 ]
Simptomi Charcot-Marie-Toothova bolest
Tipično, prvi znakovi ove bolesti počinju se pojavljivati u djetinjstvu i adolescenciji te se postupno razvijaju tijekom života, iako se sindrom može pojaviti kasnije. Kombinacija simptoma je varijabilna, a brzina napredovanja bolesti, kao i njezina težina, nemoguće je predvidjeti.
Tipični simptomi početne faze uključuju povećani opći umor; smanjeni tonus (slabost) mišića stopala, gležnjeva i potkoljenica; nedostatak refleksa. To otežava kretanje stopala i dovodi do disbazije (poremećaja hoda) u obliku višeg podizanja nogu, često s čestim spoticanjem i padovima. Znakovi Charcot-Marie-Toothove bolesti kod malog djeteta mogu uključivati izraženu nespretnost i poteškoće u hodanju koje nisu karakteristične za dob, povezane s bilateralnim spuštanjem stopala. Karakteristične su i deformacije stopala: visoki svod (šuplje stopalo) ili teška ravna stopala, zakrivljeni (čekićasti) prsti.
U slučaju hodanja na prstima na pozadini mišićne hipotonije, neurolog može posumnjati da dijete ima CMT tipa 4, kod kojeg djeca možda neće moći hodati do adolescencije.
Kako bolest napreduje, atrofija mišića i slabost šire se na gornje ekstremitete, što otežava izvođenje fine motorike i obavljanje normalnih zadataka rukama. Smanjeni taktilni osjeti i sposobnost osjećanja topline i hladnoće, kao i utrnulost u stopalima i rukama, ukazuju na oštećenje aksona senzornih živaca.
Kod Charcot-Marie-Toothove bolesti tipa 3 i 6, koja se manifestira u djetinjstvu, opažaju se senzorna ataksija (poremećena koordinacija pokreta i ravnoteže), trzanje mišića i tremor, oštećenje facijalnog živca, atrofija vidnog živca s nistagmusom i gubitak sluha.
U kasnijim fazama može doći do nekontroliranog tremora (tresenja) i čestih grčeva mišića; problemi s kretanjem mogu dovesti do razvoja boli: mišićne, zglobne, neuropatske.
Komplikacije i posljedice
Charcot-Marie-Toothova bolest može imati komplikacije i posljedice kao što su:
- češća uganuća i prijelomi;
- kontrakture povezane sa skraćivanjem periartikularnih mišića i tetiva;
- skolioza (zakrivljenost kralježnice);
- problemi s disanjem – kada su oštećena živčana vlakna koja inerviraju mišiće dijafragme:
- gubitak sposobnosti samostalnog kretanja.
Dijagnostika Charcot-Marie-Toothova bolest
Dijagnoza uključuje klinički pregled, anamnezu (uključujući obiteljsku anamnezu), neurološki i sistemski pregled.
Testovi se provode kako bi se provjerio opseg pokreta, osjetljivost i tetivni refleksi. Živčana vodljivost može se procijeniti instrumentalnom dijagnostikom - elektromiografijom ili elektroneuromiografijom. Mogu biti potrebni i ultrazvuk ili magnetska rezonancija. [ 6 ]
Genetsko ili DNK testiranje za otkrivanje najčešćih genetskih mutacija koje uzrokuju karcinogensku melanocitnu bolest (CMT) u uzorku krvi je ograničeno jer DNK testovi trenutno nisu dostupni za sve vrste bolesti. Za više informacija pogledajte Genetsko testiranje.
U nekim slučajevima se izvodi biopsija perifernog živca (obično suralnog živca).
Diferencijalna dijagnoza
Diferencijalna dijagnoza uključuje druge periferne neuropatije, Duchenneovu mišićnu distrofiju, mijelopatske i mijasteničke sindrome, dijabetičku neuropatiju, mijelopatije kod multiple i amiotrofične lateralne skleroze, Guillain-Barréov sindrom, traumu peronealnog živca i njegovu atrofiju (uključujući i priklještenje između lumbalnih diskova kralježnice), oštećenje malog mozga ili talamusa, kao i nuspojave kemoterapije (tijekom liječenja citostaticima poput vinkristina ili paklitaksela). [ 7 ]
Tko se može obratiti?
Liječenje Charcot-Marie-Toothova bolest
Danas liječenje ove nasljedne bolesti uključuje terapiju vježbanjem (usmjerenu na jačanje i istezanje mišića); radnu terapiju (koja pomaže pacijentima sa slabošću mišića u rukama); i korištenje ortopedskih pomagala za olakšavanje hodanja. Ako je potrebno, propisuju se lijekovi protiv bolova ili antikonvulzivi. [ 8 ]
U slučajevima teških ravnih stopala može se izvesti osteotomija, a u slučaju deformacije pete indicirana je kirurška korekcija – artrodeza. [ 9 ]
U tijeku su istraživanja i genetske komponente bolesti i metoda njezinog liječenja. Upotreba matičnih stanica, određenih hormona, lecitina ili askorbinske kiseline još nije dala pozitivne rezultate.
No zahvaljujući nedavnim istraživanjima, u bliskoj budućnosti bi se doista moglo pojaviti nešto novo u liječenju Charcot-Marie-Toothove bolesti. Tako francuska tvrtka Pharnext od 2014. razvija, a od sredine 2019. i klinička ispitivanja lijeka PXT3003 za liječenje CMT tipa 1 u odraslih, koji potiskuje povećanu ekspresiju gena PMP22, poboljšava mijelinizaciju perifernih živaca i ublažava neuromuskularne simptome.
Specijalisti medicinske tvrtke Sarepta Therapeutics (SAD) rade na stvaranju genske terapije za Charcot-Marie-Toothovu bolest tipa 1. Ova terapija će koristiti bezopasni adeno-asocirani virus (AAV) iz roda Dependovirus s linearnim jednolančanim genomom DNA, koji će u tijelo prenijeti gen NTF3, kodirajući protein neurotrofin-3 (NT-3) neophodan za funkcioniranje Schwannovih živčanih stanica.
Helixmith će do kraja 2020. započeti klinička ispitivanja genske terapije Engensis (VM202) razvijene u Južnoj Koreji za liječenje mišićnih simptoma kod CMT tipa 1. [ 10 ]
Prevencija
Prevencija CMT-a može biti genetsko savjetovanje budućih roditelja, posebno ako netko u paru ima obiteljsku anamnezu bolesti. Međutim, identificirani su slučajevi de novo točkastih mutacija gena, odnosno u odsutnosti bolesti u obiteljskoj anamnezi.
Tijekom trudnoće, vjerojatnost Charcot-Marie-Toothove bolesti kod budućeg djeteta može se provjeriti biopsijom korionskih resica (od 10. do 13. tjedna trudnoće), kao i analizom amnionske tekućine (u 15.-18. tjednu).
Prognoza
Općenito, prognoza za različite vrste Charcot-Marie-Toothove bolesti ovisi o kliničkoj težini, ali u svim slučajevima bolest sporo napreduje. Mnogi pacijenti imaju invaliditet, iako to ne smanjuje očekivano trajanje života.