
Nasljedni nefritis (Alport sindrom) kod djece
Posljednji pregledao: 23.04.2024

Svi iLive sadržaji medicinski se pregledavaju ili provjeravaju kako bi se osigurala što je moguće točnija činjenica.
Imamo stroge smjernice za pronalaženje izvora i samo povezujemo s uglednim medijskim stranicama, akademskim istraživačkim institucijama i, kad god je to moguće, medicinski pregledanim studijama. Imajte na umu da su brojevi u zagradama ([1], [2], itd.) Poveznice koje se mogu kliknuti na ove studije.
Ako smatrate da je bilo koji od naših sadržaja netočan, zastario ili na neki drugi način upitan, odaberite ga i pritisnite Ctrl + Enter.
Nasljedni nefritis (alportov sindrom) - genetski određena nisu imunološki naslijeđena glomerulopatija pokazuje Hematurija (ponekad proteinurija), progresivno opadanje funkcije bubrega u razvoj kronične insuficijencije bubrega je često povezan s Senzorineuralna gluhoća i slabovidnih osoba.
Prvi put je bolest 1902. Godine opisala LGGuthrie, koja je promatrala obitelj u nekoliko generacija od kojih je promatrana hematurija. Godine 1915. članovi iste obitelji AFHurst opisuju razvoj uremije. Godine 1927. Alport je najprije identificirao gluhoću kod nekoliko rođaka s hematurijom, a tijekom 50-ih godina prošlog stoljeća opisane su ozljede oka u takvoj bolesti. Godine 1972., u bolesnika s nasljednom hematurijom, morfološki ispitivanim bubrežnim tkivom, Hinglais et al. Otkrivena neravna ekspanzija i delaminacija glomerularnih bazalnih membrana. Godine 1985. Identificirana je genetska osnova nasljednog nefritisa - mutacija u genu tipa IV kolagena (Fiengold et al., 1985).
Istraživanje genetske prirode bolesti omogućilo je zaključivanje da su razlike u fenotipskim manifestacijama nasljednog nefritisa (s ili bez gubitka sluha) zbog stupnja ekspresije mutantnog gena. Tako se sada sve kliničke varijante smatraju manifestacijama jedne bolesti, a pojam "nasljedni nefritis" sinonim je pojmom "Alportov sindrom".
Prema epidemiološkim studijama, nasljedni nefritis pojavljuje se na učestalosti od 17 na 100.000 djece.
Uzroci Alportovog sindroma
Genetska osnova bolesti je mutacija u genu a-5 kolagenskog lanca tipa IV. Ova vrsta univerzalan za bazalnih membrana, bubrega kohlearni uređaja, leća kapsule, mrežnice i rožnice koji je prikazan u studijama monoklonska protutijela protiv frakcije kolagena. Nedavno, ukazuju na mogućnost korištenja DNA probe za prenatalnu dijagnozu nasljednog nefritisa.
Naglašena je važnost testiranja svih članova obitelji pomoću DNK sondi kako bi se identificirali nositelji mutantnog gena, što je od velike važnosti u provođenju medicinskog genetičkog savjetovanja obitelji s ovom bolešću. Međutim, do 20% obitelji nema srodnika s bubrezima, što upućuje na visoku incidenciju spontanih mutacija u abnormalnom genu. Većina pacijenata s nasljednim nefritisom u obitelji ima pojedince s bubrežnim bolestima, gubitkom sluha i patologijom vida; srodnih brakova između ljudi koji imaju jedan ili više predaka, budući da brak srodnih pojedinaca povećava vjerojatnost dobivanja istih gena od oba roditelja. Utvrđeni su autosomni dominantni i autosomni recesivni i dominantni, povezani s X kromosomom prijenosa.
Djeca imaju veću vjerojatnost razlikovati tri varijante nasljednog nefritisa: Alportov sindrom, nasljedni nefritis bez gubitka sluha i benigne hematurije u obitelji.
Alportov sindrom - nasljedni nefritis s oštećenjem sluha. Temelj je kombinirani nedostatak strukture kolagena bazalne membrane glomerula bubrega, struktura uha i oka. Gen klasičnog Alport sindroma nalazi se na mjestu 21-22 q dugog kraka X kromosoma. U većini slučajeva naslijedio je dominantni tip povezan s X kromosomom. S tim u vezi, kod muškaraca, Alportov sindrom je teži, jer u žena funkcija mutantnog gena nadoknađuje zdrava alela drugog, netaknutog kromosoma.
Genetska osnova razvoja nasljednog nefritisa su mutacije u genima alfa lanaca tipa IV kolagena. Je poznat kao šest-lance tipa IV kolagena G: A5 i A6 gena lanaca (Sol4A5 i Sol4A5) se nalaze na dugom kraku kromosoma X u 21-22q zoni; geni a3- i a4-lanaca (Co4A3 i Co4A4) - na 2. Kromosomu; geni a1- i a2-lanaca (Co4A1 i Co4A2) - na 13. Kromosomu.
U većini slučajeva (80-85%), povezanost tipa bolesti s X povezana je s oštećenjem Co4A5 gena zbog brisanja, točkastih mutacija ili poremećaja spajanja. Trenutno se nalaze više od 200 mutacija gena Kol4A5, odgovorne za kršenje sinteze a5 lanaca kolagena tipa IV. U ovoj vrsti nasljeđa, bolest se očituje kod djece obaju spolova, ali kod dječaka to je teže.
Mutacije u lokacijama gena Co4A3 i Co4A4, odgovorne za sintezu a3 i a4 - lanaca tipa IV kolagena, nasljeđuju se autosomno. Prema istraživanju, autosomno dominantni tip nasljeđivanja zapažen je u 16% slučajeva nasljednog nefritisa, autosomnog recesivnog - u 6% bolesnika. Postoji oko 10 mutacija gena Co4A3 i Co4A4.
Rezultat mutacija je kršenje procesa montaže tipa IV kolagena, što dovodi do poremećaja u njegovoj strukturi. Kolagen tipa IV je jedna od glavnih komponenata glomerularne bazalne membrane, kohlearnog aparata i očne leće, koji se detektiraju patologije kod nasljednog nefritisa klinici.
Kolagen tipa IV, dio glomerularne membrane, u osnovi sastoji od dva lanaca a1 (IV) i jedan a2 (IV) lanac, a također sadrži A3, A4, A5 lanac. Najčešće kada X-vezani naslijeđe Sol4A5 mutacije uz nedostatak a3, a4 i A6 A5 lanci kolagena tipa strukture IV, a broj O1 i a2 lanaca u glomerularne bazalna membrana povećava. Mehanizam ovog fenomena je nejasan, pretpostavlja se da je uzrok posttranskripcijskih promjena u mRNA.
Nedostatak a3, a4, i A5 lanci tipa strukture kolagena IV bazalne membrane glomerula rezultata u stanjivanje i krhkosti ranim fazama alportov sindrom koji se klinički manifestira najviše Hematurija (ponekad hematurija ili proteinuriju samo proteinurija), gubitak i lenticonus sluha. Daljnje napredovanje bolesti dovodi do zadebljanja i poremećaj bazalne permeabilnosti membrane u kasnim stadijima bolesti, na rast u tim tipovima kolagena V i VI, u manifestira porastom proteinurije i smanjenu bubrežnu funkciju.
Priroda mutacije na temelju nasljednog nefritisa uglavnom određuje njegovu fenotipsku manifestaciju. Kada X kromosoma delecije s mutacijama i istovremenu Sol4A6 Sol4A5 gena odgovornih za sintezu A5 i A6 lancima kolagena tipa IV, u kombinaciji s alportov sindrom leiomyomatosis jednjaka i genitalija. Prema ispitivanjima s Sol4A5 genskih mutacija povezanih s delecijom označeni velikim ozbiljnosti patoloških procesa, kombinacija s bubrežnim lezije izvanbubrežnim manifestacije i rani razvoj kronične insuficijencije bubrega, dok stochechnoy mutacija ovog gena.
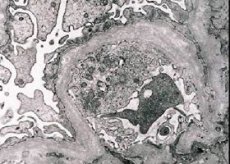
Morfološki, elektronska mikroskopija otkriva razrjeđivanje i delaminacija glomerularnih bazalnih membrana (posebno lamina densa) i prisutnost elektronski gustih granula. Oštećenje glomerula može biti neujednačeno u istom pacijentu, od minimalne fokalne lezije mezangija do glomeruloskleroze. Glomerulitis u Alportovom sindromu uvijek je imuno-negativan, što ga razlikuje od glomerulonefritisa. Karakteristični su razvoj tubularne atrofije, infiltracije limfohistiocita, prisutnost "pjenastih stanica" s inkluzijom lipida - lipofagija. S progresijom bolesti, otkriva se zadebljanje i značajno uništavanje bazalnih glomerularnih membrana.
Otkrivene su određene promjene u stanju imunološkog sustava. Pacijenti s nasljednog nefritisa smanjena razina Ig A i sklonost povećanju koncentracije krvi IgM, IgG razina može povećati u ranim stadijima bolesti i pad u kasnijim fazama. Možda je povećanje koncentracije IgM i G neka vrsta kompenzacijskog odgovora kao odgovor na deficit IgA.
Funkcionalna aktivnost T-limfocitnog sustava je smanjena; On je označio selektivnu redukciju B-limfocita, koji je odgovoran za sintezu Ig A, slomljena fagocitna imunitet vezu, uglavnom zbog sloma kcmotaksiju procesa i unutarstanične probavu neutrofila
U studiji bubrega biopsije u bolesnika s alportov sindrom elektronskim mikroskopom, ultrastrukturne promjene promatrane glomerularne bazalne membrane: stanjivanje i obrasci razdvajanja kršenje glomerularne bazalne membrane s promjenom debljine i neravnim konture. U ranim stadijima nasljednog nefritisa, defekt određuje stanjivanje i krhkost glomerularnih bazalnih membrana.
Pritiskanje glomerularnih membrana je povoljniji znak i češće kod djevojčica. Stalna elektronska mikroskopska značajka u nasljednom nefritisu je cijepanje bazalne membrane, a težina njegovog uništenja korelira s težinom procesa.

Simptomi Alportovog sindroma kod djece
Prvi simptomi Alport sindroma u obliku izoliranog urinarnog sindroma češće su otkriveni kod djece prve tri godine života. U većini slučajeva, bolest se otkriva slučajno. Mokraćni sindrom se otkriva tijekom preventivnog pregleda djeteta, prije ulaska u ustanovu za djecu ili tijekom ARVI. U slučaju pojave patologije u mokraći tijekom ARVI. U nasljednom nefritisu, za razliku od stečenog glomerulonefritisa, nema latentnog razdoblja.
U početnom stadiju bolesti, djetetovo blagostanje teško pati, karakteristična osobina je upornost i upornost urinarnog sindroma. Jedan od glavnih znakova je hematurija u različitim stupnjevima, promatrana u 100% slučajeva. Povećanje stupnja hematurije zapaženo je za vrijeme ili nakon infekcije dišnih puteva, tjelesnog napora ili nakon preventivnih cijepljenja. Proteinurija u većini slučajeva ne prelazi 1 g / dan, na početku bolesti može biti nestabilna, dok se napredak proteina povećava. Periodički, mokraćni sediment može imati leukociturija s prevagnutim limfocitima, što je povezano s razvojem međuprostornih promjena.
Kasnije, postoji kršenje djelomičnih funkcija bubrega, pogoršanje općeg stanja pacijenta: opijenost, slabost mišića, arterijska hipotenzija, često oštećenje sluha (posebno kod dječaka), ponekad oštećenje vida. Otrovanje se manifestira kao bljedilo, umor, glavobolje. U početnoj fazi bolesti, gubitak sluha u većini slučajeva otkriva se samo audiografijom. Gubitak sluha u Alportovom sindromu može se pojaviti u različitim razdobljima djetinjstva, ali najčešće je gubitak sluha dijagnosticiran u dobi od 6-10 godina. Gubitak sluha kod djece započinje na visokim frekvencijama, dostižući znatan stupanj u provođenju zraka i kostiju, prolazeći od zvuka do zvučne slušne gluhoće. Gubitak sluha može biti jedan od prvih simptoma bolesti i može prethoditi urinarni sindrom.
U 20% slučajeva bolesnici s Alportovim sindromom imaju promjene u očima. Najčešće anomalije iz leće: spherofokiya, lentikonus anterior, posterior ili mixed, različite katarakte. Kod obitelji s Alportovim sindromom postoji značajna učestalost kratkovidnosti. Brojni istraživači stalno u tim obiteljima bilježe bilateralne promjenjive promjene u obliku svijetlih bjelkastih ili žućkastih granulacija na području žutog tijela. Oni smatraju ovaj simptom kao stalni simptom, koji ima visoku dijagnostičku vrijednost u Alportovom sindromu. C. S. Chugh i sur. (1993) za oftalmološki studija pokazala pacijenti alportov sindrom smanjena oštrina vida u 66,7% slučajeva, naprijed lenticonus - 37,8%, a mrlje na mrežnici - u 22,2%, katarakte - 20%, keratokonusa - 6 , 7%.
Kod nekih djece s nasljednim nefritisom, osobito u formiranju bubrežne insuficijencije, zabilježeno je značajno zaostajanje u fizičkom razvoju. Kako napredovanje bubrežne insuficijencije razvija hipertenziju. U djece je češće otkrivena u adolescenciji iu starijim dobnim skupinama.
Karakteristična je prisutnost u bolesnika s nasljednim nefritisom raznih (više od 5-7) stigmata disembriogeneze vezivnog tkiva. Među vezivnog tkiva stigme u bolesnika s najčešći oka hypertelorism, visokog nepca, malokluzija, nenormalan oblik ušiju, zakrivljenost malog prsta na rukama „sandalevidnaya gap” na noge. Za nasljedna nefritis karakterizira jednoobraznost dizembriogeneza stigme unutar obitelji, kao i visoku učestalost njihova raspodjela u očekivanih rođaka, kroz koji se prenose bolest.
U ranim stadijima bolesti pokazala smanjenje izoliranu parcijalne bubrega: transport aminokiselina, elektroliti, koncentracije funkcija acidogeneza, daljnje promjene funkcionalno stanje kako proksimalnog i distalnog nefrona i karakter kombinaciji parcijalnih poremećaja. Smanjenje glomerularne filtracije događa se kasnije, češće u adolescentskom razdoblju. Kao što je nasljedni nefritis napreduje, anemija se razvija.
Prema tome, za nasljednog nefritisa naznačen skele od bolesti: prve faze latencije ili skrivenih kliničkih simptoma, sindroma od minimalne promjene mjehura tada nastaje postupan proces dekompenzacije sa smanjenje bubrežne funkcije s vidljive kliničke simptome (intoksikacije, astenija, razvojnih kašnjenja, anemizatsiya). Klinički simptomi se pojavljuju obično bez obzira na stratifikaciju upalne reakcije.
Nasljedni nefritis može se očitovati u različitim dobnim razdobljima, što ovisi o djelovanju gena koji je do određenog vremena u potisnutom stanju.
Klasifikacija
Postoje tri varijante nasljednog nefritisa
- Varijanta I - klinički se manifestira nefritisom s hematurijom, gubitkom sluha i oštećenjem očiju. Tijek nefritisa je progresivan uz razvoj CRF-a. Dominantna je vrsta nasljeđivanja, povezana s X kromosomom. Morfološki, postoji poremećaj strukture bazalne membrane, njegovo stanjivanje i cijepanje.
- II varijanta - klinički se manifestira nefritisom s hematurijom bez gubitka sluha. Tijek nefritisa progresivno je s razvojem kroničnog zatajenja bubrega. Dominantna je vrsta nasljeđivanja, povezana s X kromosomom. Morfološki se otkriva stanjivanje bazalne membrane glomerularnih kapilara (osobito laminadensa).
- III opcija - benigna obiteljska hematurija. Tečaj je povoljan, kronično zatajenje bubrega se ne razvija. Vrsta nasljeđivanja je autosomno dominantna ili autosomna recesivna. U autosomalnoj recesivnoj vrsti nasljeđivanja žene imaju teži tijek bolesti.
Dijagnoza Alportovog sindroma
Predloženi su sljedeći kriteriji:
- prisutnost u svakoj obitelji od najmanje dva bolesnika s nefropatijom;
- hematurija kao vodeći simptom nefropatije u probandu;
- najmanje jedan član obitelji ima gubitak sluha;
- razvoj kroničnog zatajenja bubrega kod jednog rođaka i više.
U dijagnostici raznih nasljednih i prirođenih bolesti važno mjesto pripada integriranog pristupa pregledu i iznad svega obraća pažnju na podacima dobivenim u pripremi djeteta pedigreom. Dijagnoza sindroma Alport smatra vrijedi u slučajevima kada je pacijent 3 od 4 tipične značajke: prisutnost u obiteljskoj hematurije i kroničnog zatajenja bubrega, prisutnost perceptivne sluha pacijenta, patologija otkrivanje u elektronskim mikroskopom karakterizacija biopsija znakova dekolte glomerularne bazalne membrane s promjenom debljine i neujednačene konture.
Pregled bolesnika treba uključiti kliničke genetske metode ispitivanja; usmjerena studija anamneze bolesti; opći pregled pacijenta uzimajući u obzir dijagnostičke kriterije. Naknada faza patologija može uhvatiti samo s naglaskom na takvim sindromima kao vlasništvo obiteljske povijesti, hipotenziju, više stigme dizembriogeneza mijenja sindrom mjehura. U dekompenziranom estrarenalnyh može uzrokovati simptome kao što su teške intoksikacije, astenija, retardirani fizički razvoj anemizatsiya očituje i pojačavajući s postupnim smanjenjem bubrežne funkcije. U većini bolesnika s smanjenom funkcijom bubrega opaža se smanjenje funkcije acido i aminogeneze; u 50% bolesnika primjećuje se značajno smanjenje sekrecijske funkcije bubrega; ograničavanje raspona fluktuacija optičke gustoće urina; kršenje ritma filtracije, a zatim smanjenje glomerularne filtracije. Stadij kroničnog zatajenja bubrega je dijagnosticirano prisutnošću u bolesnika za 3-6 mjeseca ili više povišenim razinama uree u serumu (više od 0,35 g / l), smanjene glomerularne filtracije do 25% od normalnih vrijednosti.
Diferencijalna dijagnoza nasljednog nefritisa mora provesti prije svega stečenog oblik hematuric glomerulonefritisa. Dobila sve akutni glomerulonefritis počinje razdoblje 2-3 tjedana nakon prethodnog infekcije, izvanbubrežnim značajke, uključujući hipertenziju sa prvih dana (u nasljednog nefritisa, s druge strane, hipotenzija), smanjeni udio glomerularne filtracije kod pojave, bez povrede djelomičnih cjevastih funkcije, dok kao i nasljedni oni su prisutni. Stečena glomerulonefritis događa s težim hematurije i proteinurije, s povećanom ESR. Dijagnostička vrijednost su tipične promjene u glomerularne bazalne membrane, karakteristična nasljedna nefritis.
Diferencijalna dijagnoza dismetabolični nefropatije provodi s kroničnim zatajenjem bubrega u obitelji identificiranog klinički monotipski bolesti bubrega, a može biti u rasponu od nefropatija pijelonefritisa do urolitijaze. Djeca često imaju pritužbe na bolove u trbuhu i povremeno mokrenjem, u sedimentu urina - oksalat.
Ako sumnjate da bi se pacijent s nasljednim nefritisom trebao poslati kako bi razjasnio dijagnozu u specijaliziranom odjelu nefrologije.
Što treba ispitati?
Kako ispitati?
Koji su testovi potrebni?
Tko se može obratiti?
Liječenje Alport sindroma
U režimu se propisuje ograničenje velikog tjelesnog napora, ostati na svježem zraku. Dijeta je vrhunska, s dovoljno sadržaja visoko kvalitetnih bjelančevina, masti i ugljikohidrata uzimajući u obzir funkciju bubrega. Od velike važnosti je identifikacija i rehabilitacija kroničnih žarišnih infekcija. Od lijekova se koriste ATP, karboksilaza, piridoksin (do 50 mg / dan), karnitin klorid. Tečajevi se održavaju 2-3 puta godišnje. Kada je hematurija propisana fitoterapija - kopriva, kopriva, kupina piva, jabuka.
U stranoj i domaćoj literaturi postoje izvještaji o liječenju prednizolonom i upotrebi citostatika. Međutim, učinak je teško suditi.
U kroničnom zatajenju bubrega koriste se hemodijaliza i transplantacija bubrega.
Nema metoda specifične (učinkovite patogene) terapije nasljednog nefritisa. Sve medicinske mjere imaju za cilj sprječavanje i usporavanje smanjenja funkcije bubrega.
Dijeta treba biti uravnotežena i visoko kalorijska, uzimajući u obzir funkcionalno stanje bubrega. U nedostatku kršenja funkcionalne države u ishrani djeteta treba biti dovoljan sadržaj proteina, masti i ugljikohidrata. U nazočnosti znakova bubrežne disfunkcije treba ograničiti količinu proteina, ugljikohidrata kalcija i fosfora, što odgađa razvoj kroničnog zatajenja bubrega.
Fizički stres bi trebao biti ograničen, djeci se savjetuje da se suzdrže od sportskih aktivnosti.
Izbjegavajte kontakt sa zaraznim pacijentima, smanjuju rizik od razvoja akutnih respiratornih infekcija. Potrebno je pročišćavati žarišta kronične infekcije. Preventivna cijepljenja za djecu s nasljednim nefritisom nisu provedena, cijepljenje je moguće samo u skladu s epidemiološkim indikacijama.
Hormonska i imunosupresivna terapija u nasljednom nefritisu je neučinkovita. Postoje naznake određenog pozitivnog učinka (smanjenje razine proteinurije i usporavanje napredovanja bolesti) s dugotrajnom primjenom inhibitora ciklosporina A i ACE već dugi niz godina.
U liječenju pacijenata koji koriste lijekove koji poboljšavaju metabolizam:
- piridoksin - 2-3 mg / kg / dan u 3 podijeljene doze tijekom 4 tjedna;
- kokaroksilaza - 50 mg intramuskularno svaki drugi dan, samo 10-15 injekcija;
- ATP - 1 ml intramuskularno svaki drugi dan, 10-15 injekcija;
- Vitamin A - 1000 U / godišnje / dan u 1 prijemu tijekom 2 tjedna;
- vitamin E - 1 mg / kg / dan u 1 prijemu tijekom 2 tjedna.
Takva terapija poboljšava opće stanje pacijenata, smanjuje cjevarsku disfunkciju, a primjenjuje se 3 puta godišnje.
Kao imunomodulator može se koristiti levamisol - 2 mg / kg / dan 2-3 puta tjedno uz intermaziju između doza od 3-4 dana.
Istraživačima hiperbarična oksigenacija ima pozitivan učinak na težinu hematurije i poremećaja bubrega.
Najučinkovitiji način liječenja nasljednog nefritisa je pravovremena transplantacija bubrega. Kada se to ne poštuje u presađivanja relapsa, u malom postotku (oko 5%) može nefritis razvoj u transplantiranog bubrega, povezane s antigenima u glomerularne bazalne membrane.
Obećavajuće područje je prenatalna dijagnoza i terapija genetskim inženjeringom. Pokusi na životinjama pokazuju veliku učinkovitost prijenosa normalnih gena odgovornog za sintezu lanaca tipa IV kolagena u bubreg, nakon čega se opaža sinteza normalnih struktura kolagena.
Pogled
Prognoza nasljednog nefritisa je uvijek ozbiljna.
Prognozno nepovoljni kriteriji za protok nasljednog nefritisa su:
- muški spolni odnos;
- rani razvoj kroničnog zatajenja bubrega kod članova obitelji;
- proteinuriju (više od 1 g / dan);
- zadebljanje glomerularnih bazalnih membrana prema mikroskopiji;
- neuritis slušnog živca;
- delecija u genu Co4A5.
Prednost dobroćudne hematurije obitelji je povoljnija.
Использованная литература