
Charcot-Marie-Tooth bolest
Posljednji pregledao: 23.11.2021

Svi iLive sadržaji medicinski se pregledavaju ili provjeravaju kako bi se osigurala što je moguće točnija činjenica.
Imamo stroge smjernice za pronalaženje izvora i samo povezujemo s uglednim medijskim stranicama, akademskim istraživačkim institucijama i, kad god je to moguće, medicinski pregledanim studijama. Imajte na umu da su brojevi u zagradama ([1], [2], itd.) Poveznice koje se mogu kliknuti na ove studije.
Ako smatrate da je bilo koji od naših sadržaja netočan, zastario ili na neki drugi način upitan, odaberite ga i pritisnite Ctrl + Enter.
Atrofija, sindrom peronealnih mišića ili Charcot-Marie-Toothova bolest cijela je skupina kroničnih nasljednih bolesti s oštećenjem perifernih živaca.
Prema ICD-10 u odjeljku bolesti živčanog sustava, šifra ove bolesti je G60.0 (nasljedna motorna i senzorna neuropatija). Također je uključen u popis siročadi.
Epidemiologija
Prema kliničkim statistikama, prevalencija svih vrsta Charcot-Marie-Tooth bolesti na 100 tisuća stanovnika iznosi 19 slučajeva (prema drugim izvorima, jedan slučaj na 2,5-10 tisuća stanovnika).
CMT tip 1 čini oko dvije trećine slučajeva (jedan slučaj na 5-7 tisuća stanovnika), a gotovo 70% njih povezano je s dupliciranjem gena PMP22. U svijetu ova vrsta bolesti pogađa više od 1,2 milijuna ljudi.
Incidencija CMT tipa 4 procjenjuje se na 1-5 slučajeva na 10 tisuća djece. [1]
Uzroci charcot-Marie-Tooth bolest
Prema klasifikaciji polineuropatskih sindroma , atrofija peronealnog (peronealnog) mišića, neuralna amiotrofija Charcot-Marie-Tooth-a ili Charcot-Marie-Toothova bolest (skraćeno CMT) odnosi se na genetski određene motoričko-senzorne polineuropatije. [2]
Odnosno, razlozi za njegovu pojavu su genetske mutacije. A ovisno o prirodi genetskih abnormalnosti, glavne vrste ili vrste ovog sindroma razlikuju se: demijelinizirajuće i aksonske. U prvu skupinu spada Charcot-Marie-Tooth-ova bolest (CMT1), koja se javlja kao rezultat umnožavanja gena PMP22 na kromosomu 17, koji kodira transmembranski periferni mijelinski protein 22. Kao rezultat, segmentna demijelinizacija aksonske ovojnice (procesi živčanih stanica) i dolazi do smanjenja brzine provođenja živca.signali. Uz to, mogu postojati mutacije nekih drugih gena.
Aksonski oblik je Charcot-Marie-Tooth bolest 2 (CMT2), koji utječe na same aksone i povezan je s patološkim promjenama u genu MFN2 na lokusu 1p36,22, koji kodira membranski protein mitofusin-2, što je neophodno za fuziju mitohondrija i stvaranje funkcionalnih mitohondrijskih mreža unutar perifernih živaca stanica. Postoji više od desetak podtipova CMT2 (s mutacijama u određenim genima).
Valja napomenuti da je sada identificirano više od stotinu gena, čija oštećenja, naslijeđena, uzrokuju razne podtipove Charcot-Marie-Tooth bolesti. Na primjer, mutacije u genu RAB7 razvijaju CMT tipa 2B; promjena gena SH3TC2 (koji kodira jedan od proteina membrane Schwannovih stanica) uzrokuje CMT tipa 4C, koji se očituje u djetinjstvu i karakterizira demijelinizacija motoričkih i osjetnih neurona (jedan i pol desetak oblika tipa 4 ovu bolest razlikuju).
Rijetki SMT tipa 3 (nazvan Dejerine-Sott sindrom), uzrokovan mutacijama gena PMP22, MPZ, EGR2 i drugih, također se počinje razvijati u ranom djetinjstvu.
Kada se CMT tip 5 pojavi u dobi od 5 do 12 godina, ne bilježi se samo motorička neuropatija (u obliku spastične parapareze donjih ekstremiteta), već i oštećenje vidnog i slušnog živca.
Slabost mišića i atrofija vidnog živca (s gubitkom vida), kao i problemi s ravnotežom, karakteristični su za CMT tip 6. A kod tipa 7 Charcot-Marie-Tooth bolesti opaža se ne samo motoričko-senzorna neuropatija, već i bolest mrežnice u obliku pigmentoznog retinitisa.
Češća SMT vezana uz X ili Charcot-Marie-Toothova bolest s tetraparezom ekstremiteta (slabljenje kretanja ruku i nogu) među muškarcima je demijelinizirajućeg tipa i smatra se rezultatom mutacije gena GJB1 na dugi krak X kromosoma, koji kodira za Connexin 32, transmembranski protein Schwannove stanice i oligodendrocite, koji regulira prijenos živčanih signala. [3]
Faktori rizika
Glavni čimbenik rizika za CMT je prisutnost ove bolesti u obiteljskoj anamnezi, odnosno u bliskoj rodbini.
Prema genetičarima, ako su oba roditelja nositelji autosomno recesivnog gena bolesti Charcot-Marie-Tooth, rizik od rođenja djeteta koje će razviti ovu bolest iznosi 25%. A rizik da će dijete nositi taj gen (ali ono samo neće imati simptome) procjenjuje se na 50%.
U slučaju nasljeđivanja vezanog uz X (kada se mutirani gen nalazi na X-ovom kromosomu žene), postoji 50% rizika da će majka taj gen prenijeti sinu, a on će razviti CMT bolest. Kada se rodi žensko dijete, bolest se možda neće dogoditi, ali kćerkini sinovi (unuci) mogu naslijediti neispravni gen - s razvojem bolesti.
Patogeneza
Kod bilo koje vrste Charcot-Marie-Tooth bolesti, njegova je patogeneza posljedica nasljedne anomalije perifernih živaca: motornih (motoričkih) i osjetnih (osjetnih).
Ako je CMT tip demijelinizirajući, tada uništavanje ili defekt mijelinske ovojnice, koja štiti aksone perifernih živaca, dovodi do usporavanja prijenosa živčanih impulsa perifernog živčanog sustava - između mozga, mišića i osjetnih organa.
Kod aksonskog tipa bolesti aksoni su izravno zahvaćeni, što negativno utječe na snagu živčanih signala, što je nedovoljno za potpunu stimulaciju mišića i osjetnih organa.
Također pročitajte:
Kako se širi Charcot-Marie-Tooth sindrom? Neispravni geni mogu se naslijediti na autosomno dominantan ili autosomno recesivan način.
Najčešće - autosomno dominantno nasljeđivanje - događa se kada postoji jedna kopija mutiranog gena (koju nosi jedan od roditelja). A vjerojatnost prijenosa CMT-a na svako rođeno dijete procjenjuje se na 50%. [4]
U autosomno recesivnom nasljeđivanju bolest zahtijeva dvije kopije neispravnog gena (po jednu od svakog roditelja koji nema znakove bolesti).
U 40-50% slučajeva dolazi do autosomno dominantne nasljedne demijelinizacije, odnosno CMT tipa 1; u 12-26% slučajeva - aksonski CMT, odnosno tip 2. I u 10-15% slučajeva opaža se nasljeđivanje vezano za X. [5]
Simptomi charcot-Marie-Tooth bolest
Obično se prvi znakovi ove bolesti počinju javljati u djetinjstvu i adolescentima te se postupno razvijaju tijekom života, iako se sindrom može osjetiti i kasnije. Kombinacija simptoma je promjenjiva, a brzinu napredovanja bolesti, kao ni njezinu težinu, nije moguće predvidjeti.
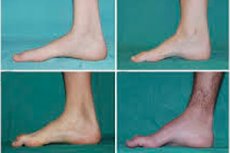
Postoje takvi tipični simptomi početne faze kao povećani opći umor; smanjeni tonus (slabost) mišića stopala, gležnjeva i potkoljenica; nedostatak refleksa. To otežava pomicanje stopala i dovodi do disbazije (poremećaja hoda) u obliku veće kote nogu, često uz često posrtanje i padanje. Znakovi bolesti Charcot-Marie-Tooth kod malog djeteta mogu biti izražena nespretnost i poteškoće u hodu, neuobičajene za dob, povezane s obostranim visećim stopalom . Karakteristične su i deformacije stopala: visoki luk (šuplje stopalo) ili jaka ravna stopala, zakrivljeni (poput čekića) prsti.
U slučaju hodanja na prstima na pozadini mišićne hipotenzije, neurolog može sumnjati da dijete ima CMT tip 4, u kojem djeca do adolescencije možda neće moći hodati.
Kako napreduju, atrofija i slabost mišića šire se na gornje ekstremitete, otežavajući finu motoriku i normalne aktivnosti ruku. Smanjenje taktilnih osjeta i sposobnost da se osjećaju toplo i hladno, kao i utrnulost stopala i ruku, ukazuje na oštećenje aksona osjetnih živaca.
S manifestiranom u djetinjstvu Charcot-Marie-Tooth bolešću tipa 3 i 6, dolazi do osjetljive ataksije (poremećena koordinacija pokreta i ravnoteže), trzanja mišića i podrhtavanja, oštećenja facijalnog živca, atrofije vidnog oka s nistagmusom, gubitka sluha.
U kasnijim fazama može doći do nekontroliranog podrhtavanja (podrhtavanja) i čestih grčeva u mišićima; problemi s kretanjem mogu dovesti do razvoja boli: mišićne, zglobne, neuropatske.
Komplikacije i posljedice
Charcot-Marie-Tooth bolest može imati komplikacije i posljedice kao što su:
- češći uganuća i prijelomi;
- kontrakture povezane sa skraćivanjem periartikularnih mišića i tetiva;
- skolioza (zakrivljenost kralježnice);
- problemi s disanjem - s oštećenjem živčanih vlakana koja inerviraju mišiće dijafragme:
- gubitak sposobnosti samostalnog kretanja.
Dijagnostika charcot-Marie-Tooth bolest
Dijagnostika uključuje klinički pregled, povijest (uključujući obiteljsku povijest), neurološki i sistemski pregled.
Ispitivanja se provode radi provjere opsega pokreta, osjetljivosti i tetivnih refleksa. Provođenje živca može se procijeniti instrumentalnom dijagnostikom - elektromiografijom ili elektroneuromiografijom . Također će biti potreban ultrazvuk ili MRI. [6]
Genetska ili DNK dijagnostika za identificiranje najčešćih genetskih mutacija koje uzrokuju CMT na uzorku krvi su ograničene, jer DNK testovi trenutno nisu dostupni za sve vrste CMT-a. Za detalje pogledajte - Genetska istraživanja
U nekim se slučajevima radi biopsija perifernog živca (obično gastrocnemius).
Diferencijalna dijagnoza
Diferencijalna dijagnoza provodi se s ostalim perifernim neuropatijama, Duchenneovom mišićnom distrofijom, mijelopatijskim i miasteničnim sindromima, dijabetičkom neuropatijom, mijeloaptijama u slučaju multiple i amiotrofične lateralne skleroze, Guillain-Barréovim sindromom, traumom peronealnog živca i atrofijom diska kralježnice ), oštećenja malog mozga ili talamusa, kao i nuspojave kemoterapije (kada se liječe citostaticima kao što su vinkristin ili paklitaksel). [7]
Tko se može obratiti?
Liječenje charcot-Marie-Tooth bolest
Danas se liječenje ove nasljedne bolesti sastoji u fizioterapijskim vježbama (usmjerenim na jačanje i istezanje mišića); radna terapija (koja pomaže pacijentima s slabošću mišića u rukama); pomoću ortopedskih pomagala za olakšavanje hodanja. Ako je potrebno, uzmite lijekove protiv bolova ili antikonvulzive. [8]
U slučajevima izraženih ravnih stopala može se izvršiti osteotomija, a u slučaju deformacije peta indicirana je njihova kirurška korekcija - artrodeza. [9]
U tijeku su istraživanja kako genetske komponente bolesti, tako i metoda liječenja. Korištenje matičnih stanica, nekih hormona, lecitina ili askorbinske kiseline još nije dalo pozitivne rezultate.
No, zahvaljujući najnovijim istraživanjima, u bliskoj će se budućnosti moglo zaista pojaviti novo u liječenju Charcot-Marie-Tooth bolesti. Dakle, od 2014. Francuska tvrtka Pharnext razvija, a od sredine 2019. Klinička ispitivanja lijeka PXT3003 za liječenje CMT tipa 1 kod odraslih, suzbijajući povećanu ekspresiju gena PMP22, poboljšavajući mijelinizaciju perifernih živaca i slabljenje neuromuskularnih simptoma.
Specijalisti medicinske tvrtke Sarepta Therapeutics (SAD) rade na genetskoj terapiji za bolest Charcot-Marie-Tooth tipa 1. Ova će se terapija koristiti bezopasnim adeno-povezanim virusom (AAV) roda Dependovirus s linearnim jednolančanim DNA genomom, koji će u tijelo nositi gen NTF3, koji kodira protein neurotropin-3 (NT-3) neophodan za funkcioniranje Schwannovih živčanih stanica.
Do kraja 2020. Godine Helixmith započinje klinička ispitivanja genske terapije Engensis (VM202) razvijene u Južnoj Koreji za liječenje mišićnih simptoma kod tipa 1 CMT. [10]
Prevencija
Prevencija CMT-a može biti genetsko savjetovanje budućih roditelja, posebno ako netko od bračnog para ima ovu bolest u obitelji. Međutim, identificirani su slučajevi de novo mutiranja genskih točaka, odnosno u nedostatku bolesti u obiteljskoj anamnezi.
Tijekom trudnoće, uzorkovanje horionskih resica (od 10 do 13 tjedana trudnoće), kao i analiza plodne vode (u 15-18 tjednu), omogućuje vam provjeru vjerojatnosti pojave Charcot-Marie-Tooth bolesti u nerođenog djeteta.
Prognoza
Općenito, prognoza za različite vrste Charcot-Marie-Tooth bolesti ovisi o kliničkoj težini, ali u svakom slučaju bolest polako napreduje. Mnogi pacijenti imaju invaliditet, iako to ne smanjuje očekivani životni vijek.